高效液相色谱法测定海水鱼中嘌呤含量
2020-05-24李婷婷任丽琨王当丰宋敏杰于海凤励建荣牟伟丽黄建联
李婷婷 任丽琨 王当丰 宋敏杰 于海凤 励建荣*谢 晶 沈 琳 牟伟丽 黄建联
(1 大连民族大学生命科学学院 辽宁大连116600 2 渤海大学食品科学与工程学院 辽宁省食品安全重点实验室 辽宁锦州121013 3 上海海洋大学食品学院 上海201306 4 大连东霖食品股份有限公司 辽宁大连116000 5 蓬莱京鲁渔业有限公司 山东烟台265600 6 福建安井食品股份有限公司 福建厦门361022)
痛风是日常生活中常见的慢性非传染型疾病,主要是由人体嘌呤代谢紊乱及尿酸排泄受阻所致[1]。近年来,随着研究者对痛风的深入研究,人们发现痛风除与遗传有关外,饮食也起着重要的作用[2]。由于我国居民生活水平的提高及膳食结构的改变,我国痛风患病率逐年上升。高秀林等[3]对我国93 例痛风患者临床资料进行分析,发现沿海地区痛风发病率高于内陆地区,推测可能与膳食结构有关。沿海地区鱼、虾、贝类资源丰富,丰富的海产资源为提高当地居民的营养水平做出了巨大贡献,然而,海产品中嘌呤含量较高,过多的摄入会导致人体嘌呤含量增多,尿酸水平升高,最终诱发痛风。了解海水鱼中嘌呤含量及分布,可为消费者尤其是痛风患者选择合适的食品提供科学指导。
现有的嘌呤检测方法主要有高效液相色谱法[4]、气相色谱法[5]、毛细管电泳法[6]以及离子对交换法[7]等。在众多方法中,鉴于高效液相色谱具有简便、快速、灵敏、准确的特点[8],已在嘌呤检测领域获得广泛的应用。然而,由于食品种类不同,所以无法建立完善、统一的高效液相色谱法的样品前处理方法。本研究旨在建立海水鱼中嘌呤含量检测的高效液相色谱法,检测常见的海水中的鱼嘌呤,完善海水鱼嘌呤数据,为痛风及高尿酸血症患者合理地食用海水鱼提供理论依据。
1 材料与方法
1.1 原料及药品
新鲜鳕鱼、美国红鱼、金仓鱼、海鲈鱼、黄花鱼、踏板鱼及鲜活大菱鲆,购于锦州水产市场;腺嘌呤、鸟嘌呤、次黄嘌呤、黄嘌呤(色谱级,纯度≥99.0%),aladdin 公司;甲醇(色谱纯),aladdin 公司;四丁基氢氧化铵(色谱纯),aladdin 公司;冰乙酸(色谱纯),aladdin 公司;三氟乙酸(色谱纯),山东西亚化学股份有限公司;甲酸(分析纯),天津市津东天正精细化学试剂厂;高氯酸(分析纯),天津市风船化学试剂科技有限公司。
1.2 仪器及设备
MS105DU 型电子分天平,瑞士梅特勒-托利多有限公司;LC-2030 型岛津高效液相色谱仪,日本岛津公司;DELTA 320 型pH 计,瑞士Mettler toledo 公司;UV-2700 型紫外-可见分光光度计,日本岛津公司;SK6210HP 型超声清洗器,上海科导超声仪器有限公司;Biofuge Stratos 型冷冻高速离心机,美国Thermo 公司;EYELAN-1100 型真空旋转蒸发仪,东京理化公司;JYS-A800 型绞肉机,九阳股份有限公司;DK-8D 型电热恒温水槽;MiliQ 型超纯水系统,美国Millipore 公司。
1.3 方法
1.3.1 色谱条件
1.3.1.1 流动相的选择
①参考程庆红等[9]的色谱条件并稍作修改。色谱柱为Agilent ZORBAX Eclipse XDB-C18(4.6 mm×250 mm,5 μm),流动相为甲醇-水(V/V=5/95),流速1.0 mL/min,柱温30 ℃,检测波长254 nm,进样量20 μL。探究不同甲醇浓度(甲醇∶水体积比分别为2∶98,7∶93,10∶90)对分离结果的影响。
②采用刘镇等[10]、刘桂英等[11]的色谱条件。具体方法如下:色谱柱为Agilent ZORBAX Eclipse XDB-C18(4.6 mm×250 mm,5 μm),流动相为0.007 mol/L KH2PO4-H3PO4(pH=4.0),流速1.0 mL/min,柱温25 ℃,检测波长254 nm,进样量20 μL。
③使用曲欣等[12]检测食品中嘌呤含量的色谱条件,色谱柱为Agilent ZORBAX Eclipse XDBC18(4.6 mm×250 mm,5 μm),流动相为水-甲醇-冰乙酸-10%四丁基氢氧化铵(V/V/V/V=879/100/15/6),流速1.0 mL/min,柱温30 ℃,检测波长254 nm,进样量10 μL。探究最佳离子对试剂浓度。
1.3.1.2 流速的确定 使用选定的最佳流动相,保持其它色谱条件不变,分别以流速0.8,1.0,1.2,1.4 mL/min 对4 种嘌呤进行分离,确定4 种嘌呤分离的最适流速。
1.3.1.3 检测波长的确定 配制质量浓度为10 mg/L 的4 种嘌呤溶液(腺嘌呤、鸟嘌呤、次黄嘌呤、黄嘌呤),在200~400 nm 范围对其进行全光谱扫描,找出最适检测波长。
1.3.2 样品处理 将海水鱼去鳞,取鱼皮、腹部鱼肉、背部鱼肉、鱼眼睛、内脏,用绞肉机将其绞碎,分类保存。
1)准确称取0.2000 g 样品加入8 mL 超纯水,超声提取150 min,将提取液转移至离心管中,以10 000 r/min 离心10 min,取上清液用超纯水定容10 mL[13-14],经0.45 μm 滤膜过滤备用。
2)准确称取0.2000 g 样品加入2 mL 6%高氯酸,震荡40 s 使其混合均匀,水浴60 min 并分别在30,40,50 min 时震荡40 s,提取液冰浴冷却。用氢氧化钾溶液调pH 值至7,滤去沉淀,用甲酸将pH 值调至3.6,用最终确定的流动相定容10 mL[15],经0.45 μm 滤膜过滤,备用。
3)参照Havlik J 等[16]的样品处理方法,称取0.2000 g 绞碎的样品置于50 mL 离心管中,加入5 mL 三氟乙酸及5 mL 甲酸,85 ℃水浴15 min 后迅速冷却,然后于50 ℃旋转蒸发至干,残余物用10 mL 确定的最适流动相复溶。将复溶后的液体以3 000 r/min 转速离心20 min,取上清液过0.45 μm 滤膜,备用。
1.3.3 标准曲线的制作 配制1 600 mg/L 的单一标准储备液,4 ℃冰箱保存。检测前取等体积的4种嘌呤单一标准储备液,配制质量浓度为400 mg/L 的混合标准储备液,用超纯水稀释混标,配制质量 浓 度 分 别 为300,200,100,50,10,5,1,0.5,0.1 mg/L 的系列梯度溶液,经0.45 μm 微孔滤膜过滤后进行色谱分析。以峰面积(Y)及样品质量浓度(X)作标准曲线,以3 倍信噪比(S/N)所对应的浓度为检出限(limits of detection,LOD)。
1.3.4 方法精密度 将300 mg/L 的混合标品用已确定的最佳色谱方法连续进样6 次,根据测定量计算其相对标准偏差(RSD)。
1.3.5 重复性试验 取大菱鲆背部鱼肉样品6份,每份0.2000 g,按照确定的最佳嘌呤提取方法处理样品。将6 份处理的样品用选定的色谱条件进行高效液相色谱检测,根据测定量计算其相对标准偏差(RSD),检验水解方法的重现性。
1.3.6 加标回收率 准确称取大菱鲆鱼肉样品12 份,每份0.2000 g。使用最佳嘌呤提取方法对12 份样品进行处理,按照1.3.1 节确定的最适色谱条件测定其嘌呤含量。之后,按每种嘌呤含量的0.5,1,2 倍加入嘌呤标品,配制成低、中、高3 个浓度,上机检测。每个样品测定3 次,求回收率和相对标准偏差。
2 结果与分析
2.1 优化的色谱条件
2.1.1 流动相 采用V甲醇∶V水=5∶95、0.007 mol/L KH2PO4-H3PO4(pH=4.0)及水-甲醇-冰乙酸-10%四丁基氢氧化铵(V/V/V/V=879/100/15/6)作为流动相,测定嘌呤标准品中4 种嘌呤的含量。
当以V甲醇∶V水=5∶95 为流动相时,结果如图1a 所示,4 种嘌呤中只能分离出3 种嘌呤,从左到右依次为鸟嘌呤、次黄嘌呤、腺嘌呤,无法使4 种嘌呤分离。这与程庆红等[17]对嘌呤标品的检测结果相差较大,推测可能与色谱柱型号有关。程庆红等[17]选用TC-C18色谱柱,而本文用Agilent ZORBAX Eclipse XDB-C18 色谱柱。两种色谱柱虽均为C18色谱柱,但采用的填料技术不同。前者采用多孔硅胶微球,后者使用高纯度硅胶,高纯度的硅胶填料在色谱检测中与流动相中的水相溶液亲和度高。当其流动相中水所占比重较大时,使用TCC18色谱柱可以更好地保留极性化合物。另外,调节两种流动相的体积比:甲醇∶水分别为2∶98,7∶93,10∶90,结果发现甲醇的添加量对嘌呤的分离度影响虽不大,但仍无法将4 种嘌呤完全分离。最终判定此流动相不适合多组分嘌呤分离。V甲醇∶V水=10∶90 的分离结果见图1b。

图1 嘌呤标准品的高效液相色谱图Fig.1 HPLC chromatogram of standards solution
使用刘镇等[10]、刘桂英等[11]选出的最佳流动相0.007 mol/L KH2PO4-H3PO4(pH=4.0)对4 种嘌呤进行分离,结果如图2所示,相较甲醇-水流动相,磷酸盐可使4 种嘌呤基线分离,色谱峰从左到右依次为鸟嘌呤、次黄嘌呤、黄嘌呤、腺嘌呤,鸟嘌呤及腺嘌呤峰面积较小而未达到理想分离效果。现有研究表明[18],嘌呤有极性,而KH2PO4-H3PO4极性较小,两者极性的差异导致嘌呤与流动相间结合不紧密,最终致使色谱图中峰面积较小。本文参照毛玉涛等[19]的方法向KH2PO4-H3PO4中加入少量的(5%,10%)甲醇溶液,增加流动相极性,改善与周围介质的亲和作用[20]。结果发现在磷酸二氢钾缓冲液中添加甲醇虽对4 种嘌呤的分离效果影响不大,但仍存在峰面积较小的问题。曲欣[21]在KH2PO4-H3PO4基础上添加少量甲醇,同样发现甲醇对多组分嘌呤分离度影响较小,与本试验结果相同。此外,使用纯磷酸盐作为流动,除以上峰面积较小的问题外,还存在色谱柱被盐分腐蚀的危险,使柱压升高,柱效下降,因此不建议使用。
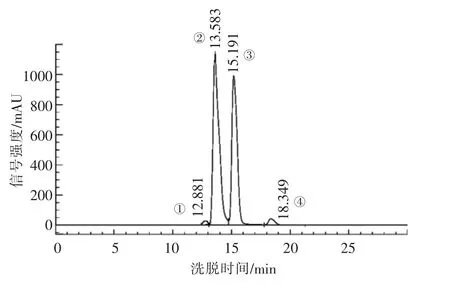
图2 嘌呤标准品的高效液相色谱图Fig.2 HPLC chromatogram of standards solution
曲欣[21]将10%四丁基氢氧化铵、冰醋酸、超纯水、甲醇以V/V/V/V=6/15/879/100 的比例混和,使腺嘌呤、鸟嘌呤、次黄嘌呤、黄嘌呤成功分离。流动相的4 种成分中甲醇用来改变流动相极性,提高分离效率。冰乙酸可以调节流动相pH,保持嘌呤活性,帮助嘌呤从固定相中分离[22],提高检测准确度。而四丁基氢氧化铵可与嘌呤生成中性离子,增强与非极性相的作用。在文献[21]的基础上探究了20%,30%,40%四丁基氢氧化铵对嘌呤分离效果的影响(见图3),结果发现当四丁基强氧化铵含量为10%时,4 种嘌呤不能基线分离且黄嘌呤有拖尾现象,可能是由于黄嘌呤中含有N 原子,N 原子与C18色谱柱上的硅醇基相互作用,当不加入离子对试剂或离子对试剂浓度较小时会产生严重的拖尾现象[23]。应提高四丁基氢氧化铵浓度。20%,30%及40%的四丁基氢氧化铵分离效果较好,无拖尾现象,保留时间较为接近,其中20%的四丁基氢氧化铵峰值较高且节约流动相。最终选择其作为流动相中四丁基氢氧化铵最适含量。
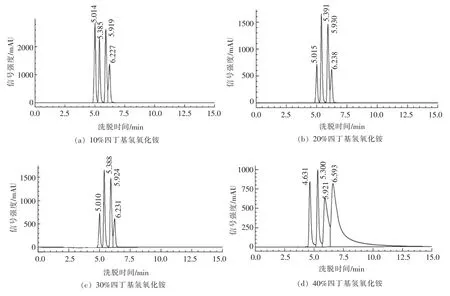
图3 不同浓度四丁基氢氧化铵对分离效果的影响Fig.3 Influence concentrtion of Tetrabutyl ammonium hydroxide on purine bases separation
2.1.2 流速 选用水-甲醇-冰乙酸-20%四丁基氢氧化铵(V/V/V/V=879/100/15/6)为流动相,保持柱温、检测波长、进样量与曲欣[21]方法一致,分别以流速0.8,1.0,1.2,1.4 mL/min 对4 种嘌呤进行分离。结果表明:使用以上流速,4 种嘌呤均在10 min 内分离,当流速为0.8 mL/min 时分离度增加,分离效果提高,峰型较好。随着流速的增大,保留时间缩短,当流速为1.4 mL/min 时色谱峰扩张降低,峰形较为尖锐,分离度下降,样品中的杂峰难以分开,降低检测准确度。高流速使柱压升高,更易损坏色谱柱,缩短其使用寿命[24]。多数研究者采用1.0 mL/min 作为检测流速,从图4可以看出,0.8 mL/min 时的分离度略大于1.0 mL/min 的分离度,有利于分离样品中的杂峰,提高检测准确度,更好地保护色谱柱。综上所述,本研究选用流速0.8 mL/min 为检测流速。
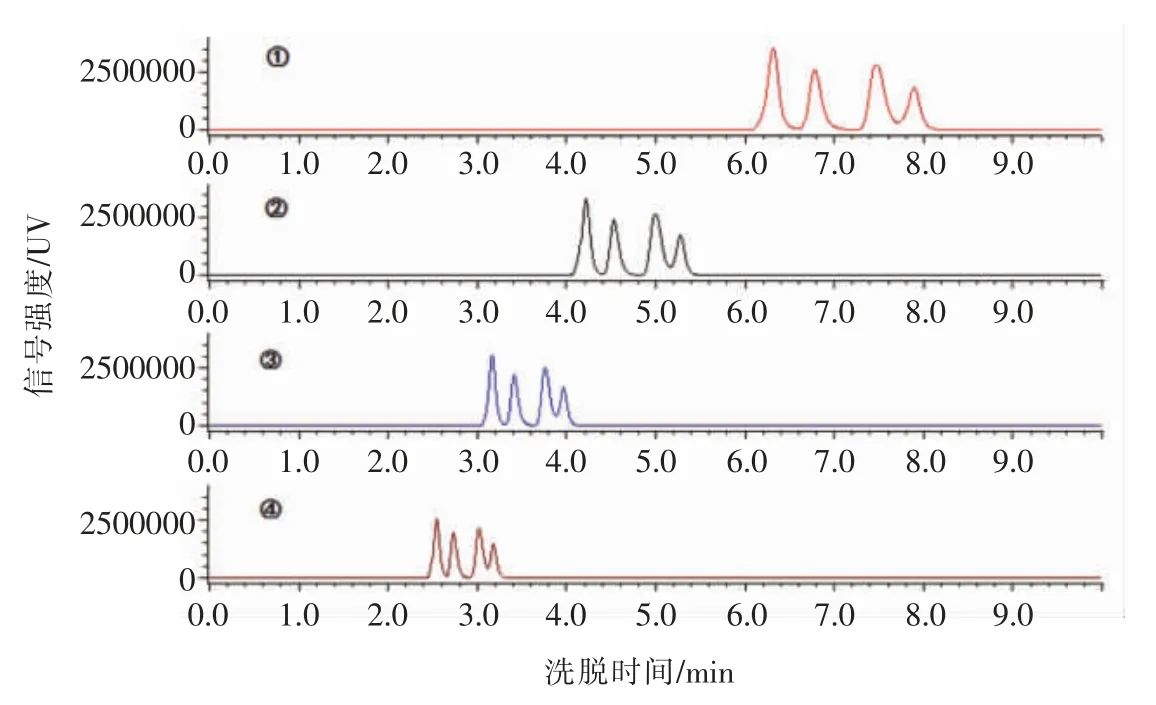
图4 不同流速下嘌呤的分离效果Fig.4 Effect of flow velocity on purine bases separation
2.1.3 检测波长 对4 种嘌呤(腺嘌呤、鸟嘌呤、次黄嘌呤、黄嘌呤)在波长200~400 nm 范围进行全光谱扫描,结果见图5。从紫外吸收光谱图上可以看出4 种嘌呤在200~300 nm 之间有吸收峰。腺嘌呤、鸟嘌呤、次黄嘌呤及黄嘌呤紫外最大吸收波长分别为267,273,254,279 nm。目前,嘌呤检测所使用的紫外波长较多,Piñeiro-Sotelo M 等[18]、Inazawa K 等[25]、刘镇[10]等分别在紫外波长255,260,254 nm 处检测食品中的嘌呤。本试验中分别选择254,260,270 nm 作为检测波长,结果差异不显著。最终本试验选用254 nm 作为检测波长。
经优化,最终选用Agilent ZORBAX Eclipse XDB-C18(4.6 mm×250 mm,5 μm)色谱柱,以水-甲醇-冰乙酸-20%四丁基氢氧化铵(V/V/V/V=879/100/15/6)为流动相,设置流速0.8 mL/min,柱温30 ℃,检测波长254 nm,进样量10 μL 为色谱条件,检测海水鱼中的嘌呤含量。
2.2 样品前处理方法
参照程庆红[13]、李楠[14]、Rong S 等[15]、Havlik J 等[9]的样品处理方法对大菱鲆鱼肉分别进行超声、高氯酸以及混合酸(V三氟乙酸∶V甲酸=1∶1)处理。采用2.1 节确定的色谱条件对处理样品的嘌呤含量进行检测。图6显示3 种处理方式的大菱鲆鱼肉样品嘌呤检测结果,3 种方式均可将4 种嘌呤从食品中提取出来。其中使用三氟乙酸-甲酸处理样品得到的嘌呤提取率最高,其次为高氯酸提取法,再次为超声提取法。这是由于高氯酸使嘌呤降解,检测峰面积减小。使用高氯酸操作繁琐、费时并伴有大量有毒氯气[26],一般不建议使用。超声提取法只用水作为提取剂,操作简单,然而嘌呤提取率较低。当使用三氟乙酸、甲酸混合提取法作为样品的处理方法时,可以很好地水解核苷和核苷酸,嘌呤提取率高,可能与甲酸对嘌呤的保护作用及三氟乙酸使嘌呤充分溶解有关[27]。本试验最终选用混合酸对样品进行处理。
2.3 标准曲线
采用2.1 节最终确定的液相色谱条件检测混合标品中的4 种嘌呤,以峰面积(Y)及样品质量浓度(X)作线性回归。线性关系及检出限结果见表1。4 种嘌呤在0.1~400 mg/L 质量浓度范围线性关系良好,相关系数(R)均高于0.9998。检出限在0.0465~0.1056 mg/L 之间。

图5 嘌呤紫外吸收光谱图Fig.5 UV absorption spectrum of purine
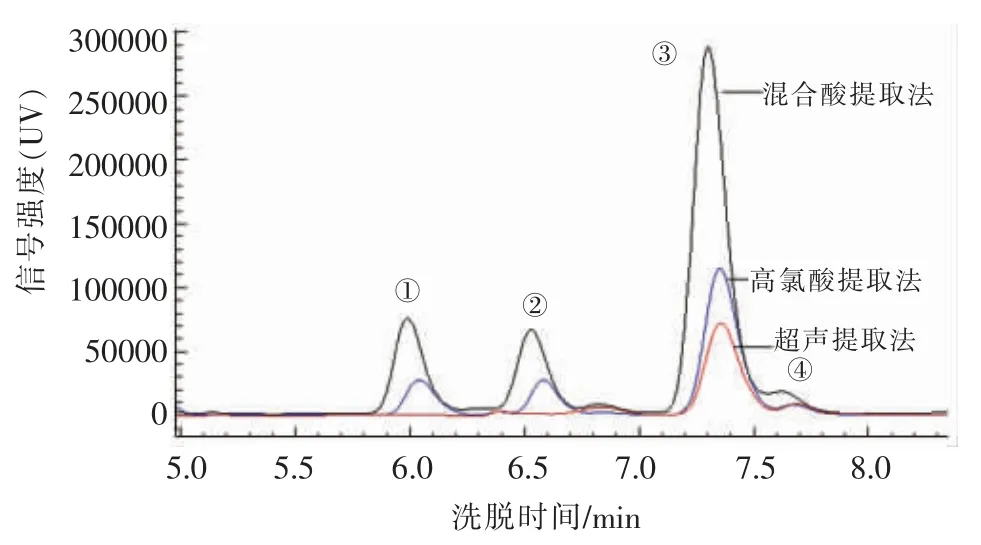
图6 不同嘌呤提取方法的样品色谱图Fig.6 Chromatogram of different purine extraction methods
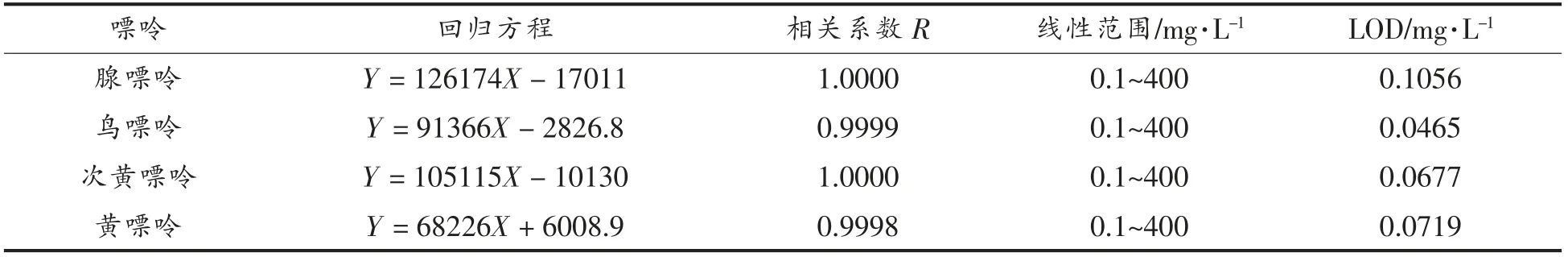
表1 线性试验结果Table1 Results of linear relation
2.4 方法精密度
相对标准偏差结果见表2,相对标准偏差均小于0.7000%,表明本试验建立的高效液相色谱检测方法精密度良好,重现性高,可应用于样品检测。
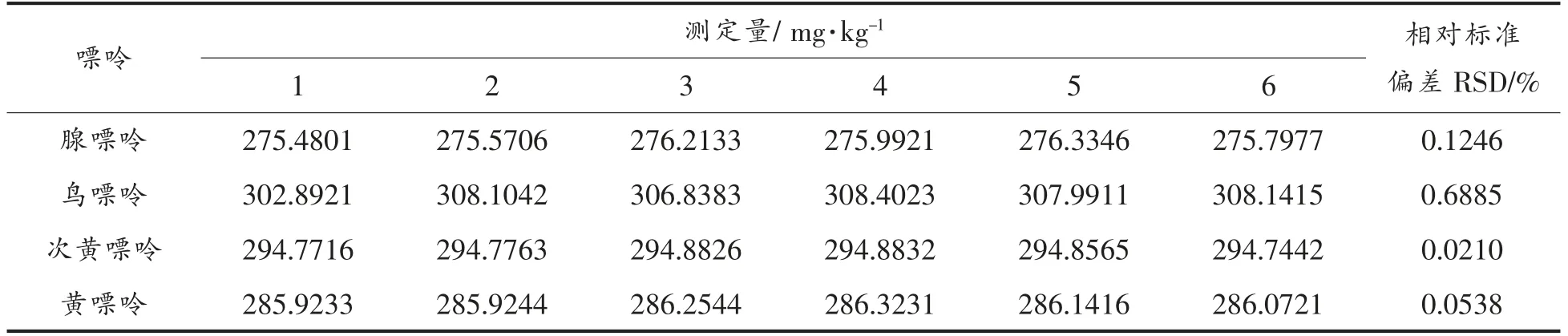
表2 HPLC 检测精密度试验结果Table2 The precision result of HPLC test
2.5 重复性
按照2.1 节、2.2 节确立的样品处理及色谱方法对大菱鲆鱼肉样品中的嘌呤进行提取和测定,结果显示:腺嘌呤、鸟嘌呤、次黄嘌呤、黄嘌呤4 种嘌呤含量的RSD 分别为1.2421%,0.9711%,0.8836%,1.9727%,重复性较好。
2.6 加标回收率
加标回收试验结果见表3,腺嘌呤、鸟嘌呤、次黄嘌呤、黄嘌呤平均回收率分别为95.9460%,94.2888%,102.9188%,95.0948%,说明采用1.3.2节样品处理方法可有效减少嘌呤损失,能较好地提取样品中的嘌呤。
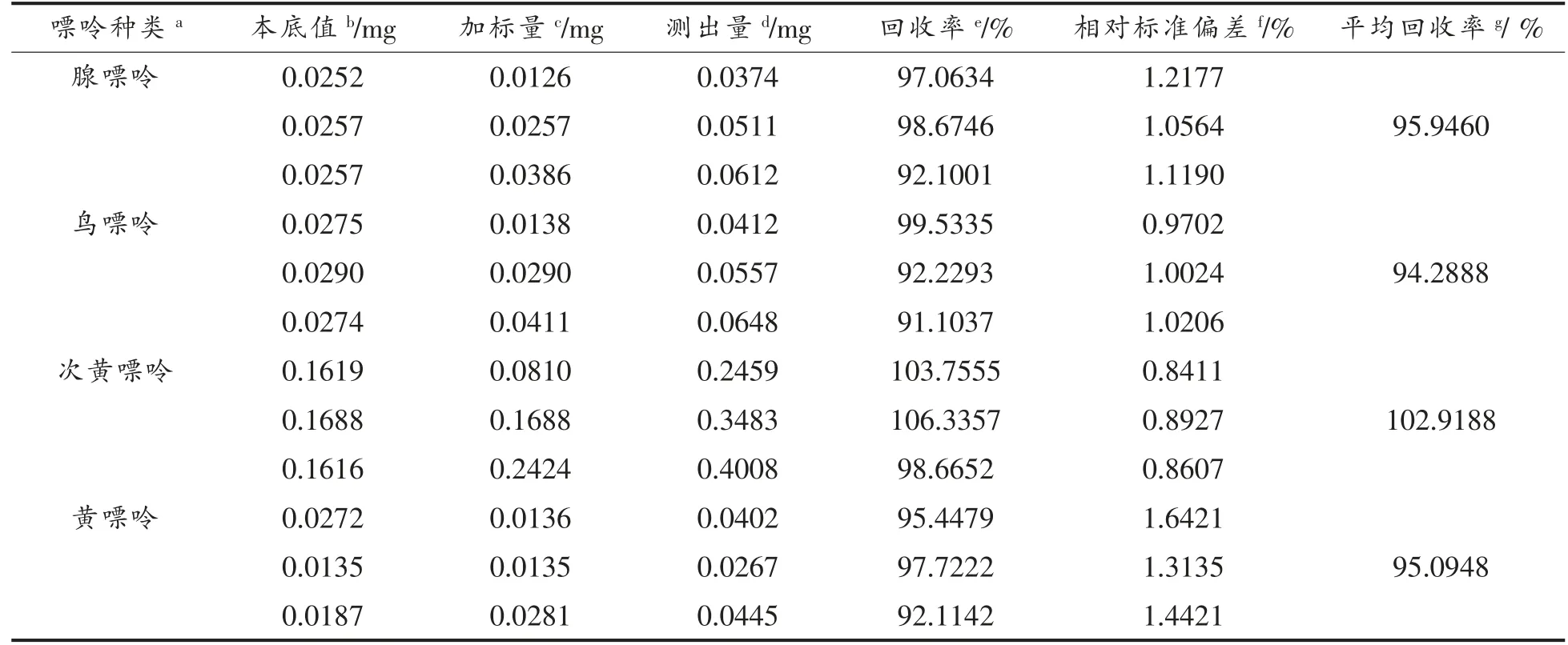
表3 加标回收试验结果表(n=3)Table3 Recovery rates of samples with added specimen(n=3)
2.7 样品嘌呤含量
将样品按照2.2 节的方法处理,并在2.1 节确定的色谱条件下检测部分海水鱼的不同可食用部分及内脏中的4 种嘌呤,每种样品平行测定3 次,结果见表4。
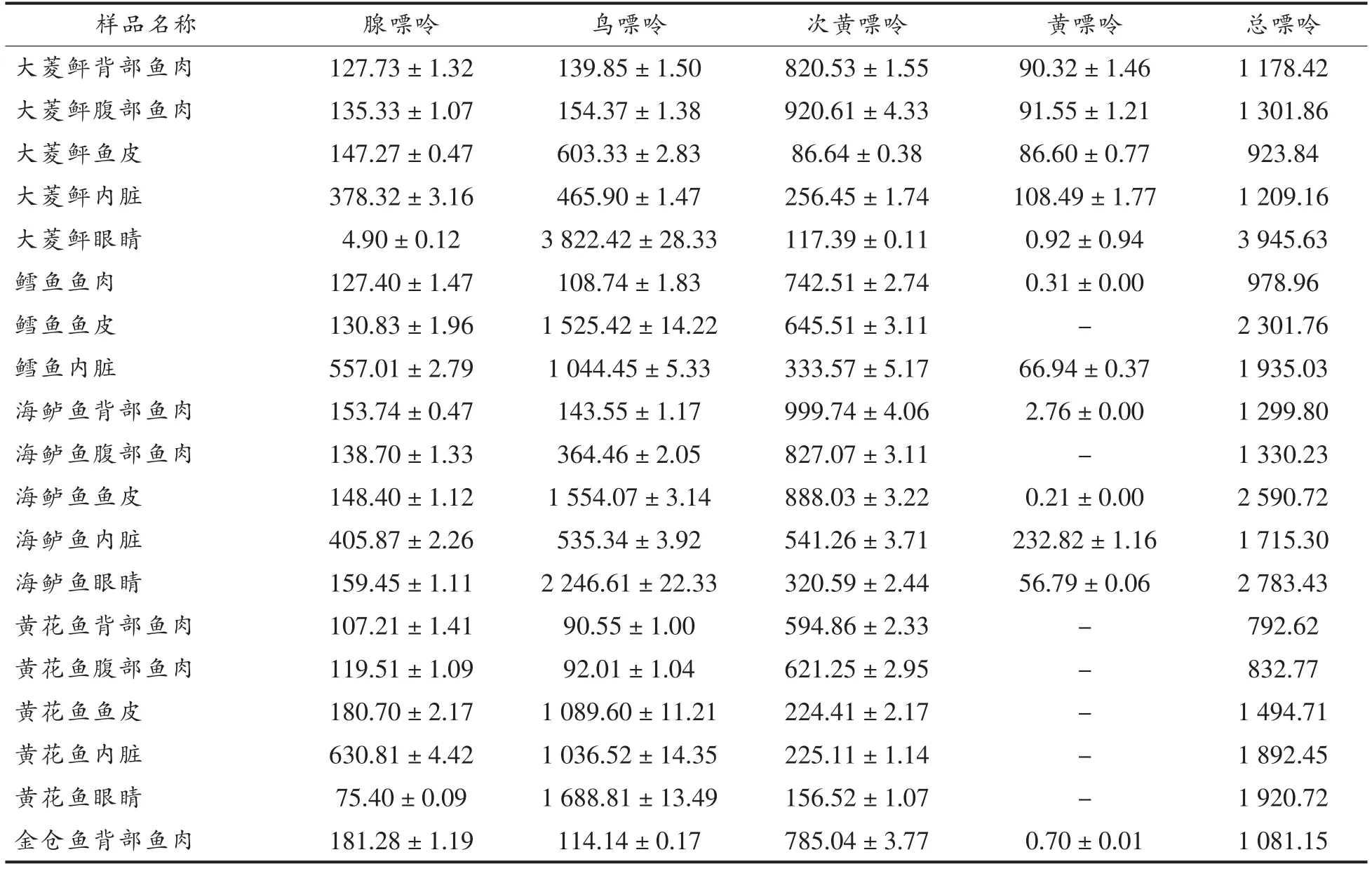
表4 样品中4 种嘌呤的含量(mg/kg)Table4 The contents of four kinds of purine in sample(mg/kg)
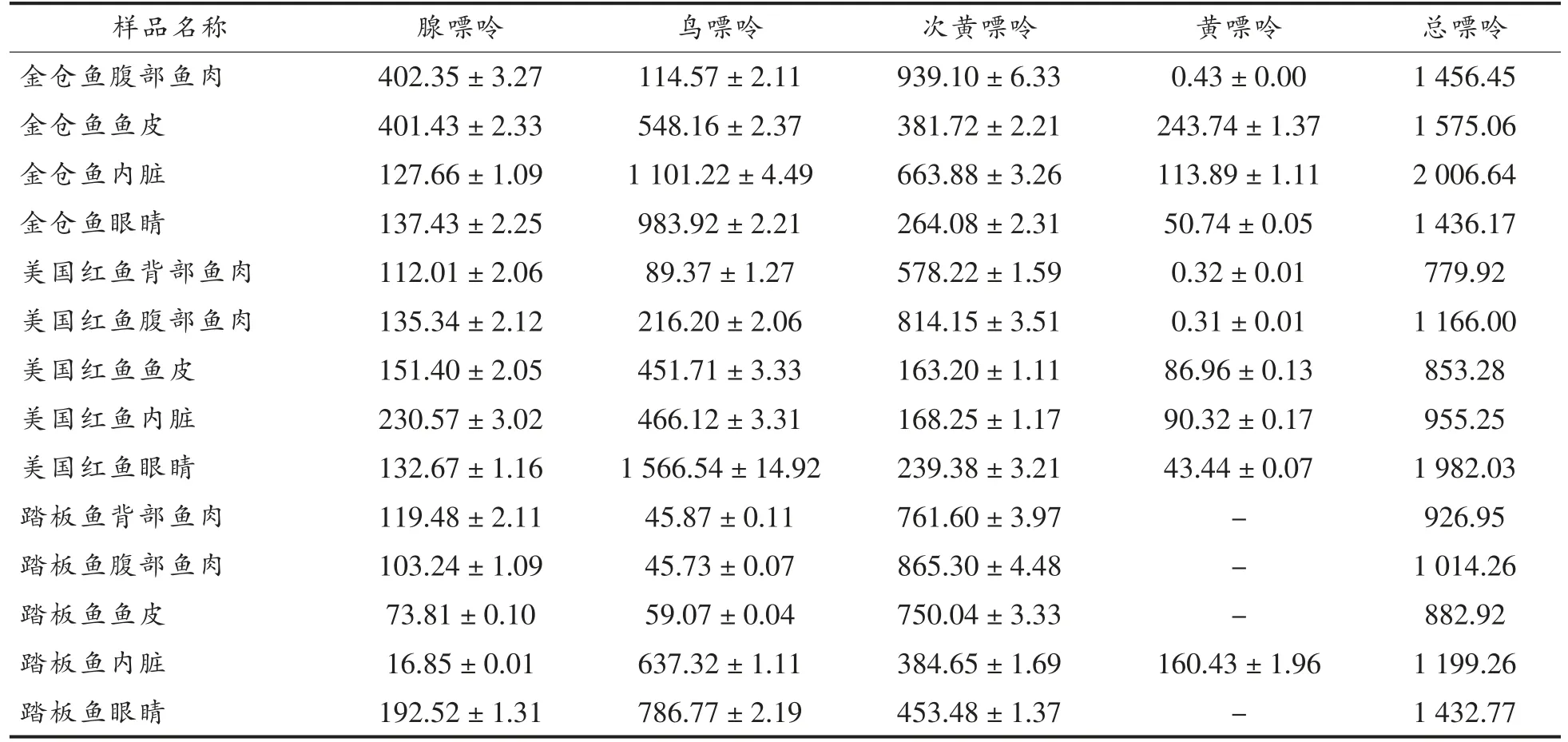
(续表4)
由表4可知,不同部位的海水鱼中嘌呤含量差异较大。鱼肉中的嘌呤含量在779.92~1 500.00 mg/kg 之间,腹部鱼肉嘌呤含量高于同种鱼类背部鱼肉的嘌呤含量。其中金仓鱼腹部鱼肉嘌呤含量高达1 456.65 mg/kg,且所有鱼肉样品中次黄嘌呤含量较高;鱼皮样品中嘌呤总量在853.25~2 600.00 mg/kg 间,不同种类的鱼,其鱼皮嘌呤总量差异较大,如海鲈鱼鱼皮的嘌呤值为2 590.72 mg/kg,美国红鱼鱼皮中嘌呤含量仅为853.28 mg/kg。此外,检测的鱼皮样品中鸟嘌呤含量均高于其它3 种嘌呤含量,为最主要的嘌呤;不同鱼内脏中鸟嘌呤含量均最高,其次为腺嘌呤和次黄嘌呤。其中黄花鱼内脏中鸟嘌呤约占总嘌呤的54.77%,未检出黄嘌呤值。海水鱼的眼睛中含有丰富的碳五烯酸和碳六烯酸[28],具有营养保健作用,成为“新晋”美食,深得消费者的喜爱,而检测的海水鱼样品中鱼眼睛的总嘌呤含量范围在1 432.77~3 945.63 mg/kg,大菱鲆鱼眼样品中总嘌呤含量高达3 945.63 mg/kg,海鲈鱼、美国红鱼总嘌呤含量次之。所有鱼眼样品中鸟嘌呤含量均较高,约占65%以上,大菱鲆样品中更是高达96.88%。
同种海水鱼嘌呤分布差异较大,所有样品中,内脏中的嘌呤含量始终高于鱼肉中的嘌呤值,这与荣胜忠[29]的研究结果一致。基体中的嘌呤大多以核苷酸的形式存在,产生上述试验结果可能是因内脏代谢率较高所致。此外,不同种类鱼的鱼皮、鱼眼睛嘌呤含量数据的差异可能与其生长环境及组织中水分含量、蛋白含量差异有关[21]。
为了预防和治疗高尿酸血症及痛风,在食用海水鱼时,建议彻底清除内脏。鱼眼睛和鱼皮虽有很高的营养价值,但要严格控制其摄入量。食用鱼肉时,建议消费者少食用金仓鱼、海鲈鱼、大菱鲆等高嘌呤含量海水鱼,合理食用美国红鱼、黄花鱼、踏板鱼和鳕鱼,且优先食用背部鱼肉。
3 结论
本试验确立了检测海水鱼中4 种嘌呤含量的高效液相色谱法。选用Agilent ZORBAX Eclipse XDB-C18(4.6 mm×250 mm,5 μm)色谱柱,以水-甲醇-冰乙酸-20%四丁基氢氧化铵(体积比879/100/15/6)为流动相,检测海水鱼中的腺嘌呤、鸟嘌呤、次黄嘌呤、黄嘌呤。经方法学验证,此方法在线性范围(0.1~400 mg/L)线性关系良好,相关系数(R)在0.9998~1.0000 之间,方法检出限为0.0465~0.1056 mg/L,高效液相色谱检测方法精密度RSD%在0.0200%~0.7000%之间。样品处理重复性试验结果表明腺嘌呤、鸟嘌呤、次黄嘌呤、黄嘌呤RSD值依次为1.2421%,0.9711%,0.8836%,1.9727%。4 种嘌呤加标回收率高于94.2888%,适用于海水鱼中4 种嘌呤的测定。应用此方法测定的部分海水鱼可食用部分及内脏的嘌呤含量数据,可为沿海地区及痛风患者提供科学健康的饮食指导。
