中枢神经系统PI3K/AKT/mTOR信号通路研究进展
2020-04-18张治楠梁丽艳连嘉惠黄芸钟正曲姗姗黄泳
张治楠 梁丽艳 连嘉惠 黄芸 钟正 曲姗姗 黄泳
1南方医科大学中医药学院(广州510515);2南方医科大学第一临床医学院(广州510515);3南方医科大学南方医院(广州510515)
PI3K/AKT/mTOR信号通路是调控细胞周期的经典通路之一,对于细胞分裂,分化,存活,肿瘤发生有重要的调控作用。越来越多的研究发现,PI3K/AKT/mTOR信号通路在中枢神经系统也扮演着各种重要角色,其异常也与中枢神经系统的许多疾病密切相关。本文就目前PI3K/AKT/mTOR信号通路的研究进展,介绍其主要组成,和在中枢神经系统的生理、病理过程中的作用,为神经系统基础研究及临床治疗提供新思路。
1 PI3K/AKT/mTOR信号转导通路的组成
1.1 PI3K为主导的启动系统 磷脂酰肌醇3⁃激酶(phosphatidylinositol 3⁃kinase,PI3K)是一种胞内磷脂酰肌醇激酶。多种生长因子和神经营养因子都能启始PI3K的激活过程,如表皮生长因子(epider⁃mal growth factor,EGF)、成纤维细胞生长因子(fi⁃broblast growth factor,FGF)、胰岛素、胰岛素样生长因子⁃1(insulin⁃like growth factor 1,IGF⁃1)、钙离子、钙调蛋白(calmodulin,CaM)[1]。当这些生长因子信号与酪氨酸激酶受体(receptor tyrosine kinases,RTKs)等跨膜受体的胞外结构域结合后,受体发生自身磷酸化,PI3K即被募集到受体磷酸化部位,催化质膜表面的磷酯酰肌醇二磷酸(phosphati⁃dylinositol⁃4,5⁃bisphosphate,PIP2)生成磷脂酰肌醇⁃3,4,5⁃三磷酸(phosphatidylinositol⁃3,4,5⁃trisphos⁃phate,PIP3),PIP3作为第二信使进一步激活下游AKT信号分子,启动PI3K/AKT通路[2]。
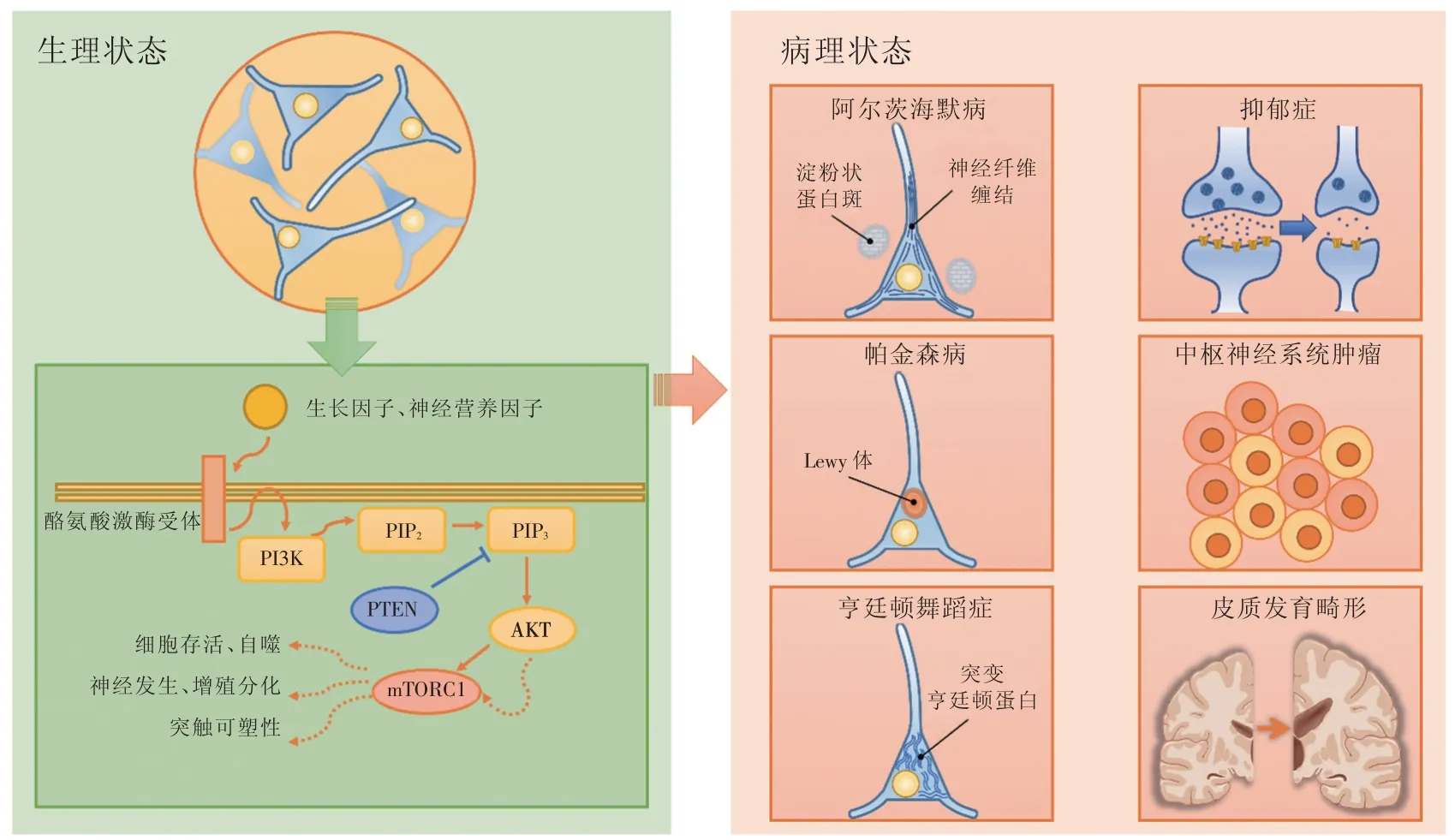
图1 PI3K/AKT/mTOR信号通路参与中枢神经系统生理、病理过程示意图Fig.1 PI3K/AKT/mTOR signaling pathway in physiological and pathological processes of central nervous system
1.2 AKT/mTOR为核心的调控节点 蛋白激酶B(protein kinase B,PKB)又称AKT,是一种胞内丝氨酸/苏氨酸(Ser/Thr)蛋白激酶。PI3K被激活在质膜上产生第二信使PIP3,PIP3与细胞内AKT结合,AKT从胞质转移至细胞膜内侧磷酸化从而激活。活化的AKT可使细胞质及细胞核内的一系列底物发生磷酸化,其中最重要的底物之一便是哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)[3]。
AKT可直接或间接磷酸化mTOR从而激活mTOR[2]。mTOR激活后,形成mTOR复合物1(mam⁃malian target of rapamycin complex 1,TORC1),是PI3K/AKT下游的核心组件[4]。mTOR通过调控多种在翻译过程中有重要作用的蛋白,如真核细胞翻译起始因子4E结合蛋白(eukaryotic translation initiation factor 4E binding proteins,eIF4EBP)和p70核糖体蛋白S6激酶(p70S6 kinase,p70S6K),启动各类蛋白质合成[5]。
1.3 PTEN为主的负调控机制 磷酸酶⁃张力蛋白基因(phosphatase and tensin homolog,PTEN)编码的蛋白PTEN是PI3K/AKT/mTOR通路的天然抑制因子。它能够将PIP3脱磷酸化变为PIP2,从而抑制PI3K/AKT通路在细胞膜上的启动程序[6]。在PTEN活性下降的情状况下,该通路活化增加,细胞表现为凋亡减少,细胞生存和增殖活跃[7]。因此PTEN是该通路最主要的负调控机制之一。
2 调控神经细胞多个生理过程
PI3K/AKT/mTOR通路激活能激活/抑制胞内各种蛋白磷酸化,调控神经细胞细胞存活/凋亡、自噬、神经发生及神经细胞增殖以及突触可塑性[8]。
2.1 细胞存活和自噬 AKT激活能够抑制细胞凋亡通路。AKT可磷酸化BCL⁃2相关细胞凋亡促进因子(bcl2⁃associated agonist of cell death,BAD),从而够抑制细胞凋亡途径的起始过程,抑制凋亡通路激活,减少细胞凋亡[9]。
总体来说,PI3K/AKT/mTOR激活抑制自噬进程,是自噬调控的主要节点[10-11]。激活的mTORC1能够催化一系列自噬启始蛋白磷酸化,从而抑制自噬的形成。此外,mTORC1也能抑制参与自噬相关蛋白合成的转录因子,减少自噬发生[12]。
2.2 神经发生及增殖分化 PI3K/AKT/mTOR通路缺乏抑制时,神经前体细胞的细胞周期间期加速,迅速进入分裂期,从而加快了神经元细胞的增殖和分化,对于神经发生及神经细胞增殖分化有重要的调节作用[13]。
2.3 突触可塑性 突触可塑性增加表现为长时程增强(long⁃term potentiation,LTP),反之则表现为长时程抑制(long⁃term depression,LTD)[14]。LTP的发生,需要激活N⁃甲基⁃D⁃天冬氨酸(N⁃methyl⁃D⁃aspartic acid,NMDA)受体,从而促进α⁃氨基⁃3⁃羟基⁃5⁃甲基⁃4⁃异恶唑丙酸(α⁃amino⁃3⁃hydroxy⁃5⁃methyl⁃4⁃isoxazolepropionic acid,AMPA)受体插入突触后膜。一方面,mTOR能增加LTP相关蛋白的表达,PI3K也能结合AMPA受体,引导其在膜上的分布[15]。NMDA受体激活导致钙离子内流和钙调蛋白激活的也能激活PI3K,相互促进。此外,AKT将磷酯酰肌醇(phosphatidylinositol,PI)固定于突触后膜,从而招募AMPA受体的对接蛋白,促进AMPA受体在突触后膜的固定[17-18]。因此,PI3K/AKT/mTOR信号通路在多个节点调控神经元突触可塑性。
3 PI3K/AKT/mTOR信号转导通路异常与神经系统疾病
3.1 阿尔茨海默病 阿尔茨海默病(Alzheimer′s disease,AD)是常见的慢性神经退行性疾病,表现为进行性记忆减退。AD发病的主要机制是淀粉状蛋白β(amyloid beta,Aβ)结合过度磷酸化的tau蛋白与神经原纤维缠结,过度沉积,导致淀粉状蛋白斑、神经纤维缠结,损伤神经元[18]。
研究[19]表明,AD与PI3K/AKT/mTOR异常激活相关。AD患者脑组织的神经原纤维缠结和过度磷酸化tau蛋白常与该通路下游的p70S6K增加同时出现[20]。且p70S6K的总量、磷酸化程度与tau蛋白总量和磷酸化tau蛋白量成正比,故tau蛋白的翻译合成与过度磷酸化均与mTOR的激活相关[21]。但p70S6K导致tau蛋白增加及过度磷酸化的机制尚不明确。有研究者提出,tau蛋白的mRNA属于5′⁃Top mRNA(5′⁃terminal oligopyrimidine tract mRNA),mTOR激活后能选择性地促进此类mRNA启动的翻译,通过促进tau的翻译来上调tau蛋白蛋白量[23]。同时,mTOR通过磷酸化作用和抑制蛋白磷酸酯酶2A(tau脱磷酸的关键酶)活性来增加tau蛋白磷酸化程度[23]。动物实验也表明3xTg AD模型小鼠与S6K1敲除小鼠杂交的后代的mTOR通路激活被抑制,其Aβ沉淀、tau蛋白磷酸化以及记忆损伤均减少[24]。且雷帕霉素、西罗莫司脂化物等mTOR抑制剂都能对抗AD模型动物脑内tau蛋白引起的神经元损伤[25⁃26]。
此外,tau蛋白也是AKT的磷酸化底物之一,AD的脑内的AKT过度激活,也是tau蛋白过度磷酸化的原因之一[27]。且AD患者脑内的真核起始因子4E(eukaryotic translation initiation factor 4E,eIF4E,mTOR下游蛋白)高出正常人100倍,mTOR可能通过激活eIF4E增加tau蛋白合成的转录过程[28]。
另一方面,PI3K/AKT/mTOR信号通路过度激活,神经元自噬水平受到抑制,无法及时清除胞内堆积的Aβ和tau蛋白,也一定程度加重了AD脑内的淀粉状蛋白斑产生和神经纤维缠结[29]。
因此,PI3K/AKT/mTOR信号通路过度激活与AD的发病密切相关。以往治疗致力于清除斑块,但这种思路并未取得突破性进展。近年来的研究提示AD的发生与神经元过度活跃相关,过度活跃的神经元不断传递错误的神经信号,导致脑内的信号处理环路损伤,促进Aβ沉积[30]。PI3K/AKT/mTOR信号通路过度激活,一方面,导致神经元的过度活跃,传递错误信号,产生损伤,同时促进Aβ沉积;另一方面,抑制自噬对Aβ的清除作用。即增加斑块形成,减少斑块清除,这无疑为临床治疗AD提供了一个新方向[25]。
3.2 帕金森病 帕金森病(Parkinson′s disease,PD)是一种中老年常见的中枢神经系统变性疾病,表现为静止性震颤、运动迟缓、肌强直和姿势步态异常,以黑质多巴胺(dopamine,DA)能神经元变性缺失和嗜酸性包涵体(Lewy体)形成为特征[31]。
Lewy体的形成主要是α⁃突触核蛋白(alpha⁃synuclein)在胞内沉积,导致神经元变性凋亡。自噬可以降解α⁃突触核蛋白,对抗Lewy体中的α⁃突触核蛋白沉淀,故PD患者脑内通常伴随自噬增加[32]。雷帕霉素可通过抑制mTOR激活自噬过程,减少α⁃突触核蛋白沉淀[33]。故抑制PI3K/AKT/mTOR信号通路能够通过促进自噬清除α⁃突触核蛋白,保护神经元,对抗PD。
但PI3K/AKT/mTOR对于PD的作用不是单向的,其激活也能对抗PD。首先,AKT或p70S6K的持续激活对PD模型小鼠的黑质纹状体神经元有保护作用[34]。敲除PTEN具有神经保护作用,促进DA神经元生长迅速[35]。mTORC1激活也能保护DA神经元,促进细胞存活而对抗PD[36]。既然激活PI3K/AKT/mTOR信号通路能够抗PD,那为何抑制mTOR也能抗PD,有研究者认为,雷帕霉素在抑制mTOR的同时,也抑制了DNA损伤诱导转录因子⁃4(DNA⁃damage⁃inducible transcript 4,DDIT4)的转录,DDIT4是mTORC1的强效抑制剂,能够通过抑制mTORC1诱导神经元凋亡,在PD发病时,该蛋白也会异常升高[37]。
多巴胺能神经元丢失是PD的关键病理改变,但目前PD的治疗多局限于补充脑内DA,在保护及恢复多巴胺能神经元上还没有有效方法,因此只能延缓病情,不能从根本上治疗PD。PI3K/AKT/mTOR通路参与PD的病理生理过程,虽然其激活会抑制自噬,但同时对多巴胺能神经元有保护作用。研究[38]也表明,激活PI3K/AKT/mTOR能促进神经元的存活,对抗PD进展。
3.3 亨廷顿病 亨廷顿病(Huntington′s disease,HD)又称亨廷顿舞蹈症,临床表现为进行性舞蹈样运动和痴呆,主要病因为第4号染色体亨廷顿蛋白(huntingtin,HTT)基因上CAG碱基序列重复次数异常增多,突变亨廷顿蛋白(mutant hunting⁃tin,mHTT)在胞浆胞核聚集,产生神经毒性,导致神经元功能受损甚至变性[37]。
mHTT能够激活PI3K/AKT/mTOR通路,减少自噬,无法清除沉积的mHTT[38]。反之,雷帕霉素通过抑制mTOR活性,减少HD模型动物mHTT聚集物,减少神经退行性病变[39]。激活HD大鼠纹状体mTOR,mHTT病理产物加快沉积,HD症状也随之加重[40]。
HD目前尚无根本性治疗手段。且HD主要由HTT基因突变产生的mHTT蛋白导致,因此降低mHTT蛋白水平是治疗亨廷顿病极具前景的治疗方法。mHTT能导致PI3K/AKT/mTOR通路异常激活,削弱了自噬对mHTT的清除作用,加速了HD的神经退行性病变,而抑制PI3K/AKT/mTOR能一定程度恢复受抑制的自噬过程,从而保护细胞,起到抗PD的作用[41]。
3.4 抑郁症 前额叶与海马是抑郁发病的两个关键脑区,近年来的研究表明抑郁的发病机制与海马、前额叶的突触可塑性改变联系密切[42⁃43]。PI3K/AKT/mTOR信号通路在多个节点调控神经元突触可塑性,抑郁状态下,该通路的活性明显下降。抑郁患者相较于正常人,其前额叶该通路激活程度下降,而且PTEN明显增加[44]。海马的该通路也被抑制,易患病人群的抑制更为明显,治疗后,通路活性恢复正常水平[45]。同时,抑郁的诱发因素,如长期摄入糖皮质激素,也会导致海马及前额叶的PTEN上调和该通路的活性明显下降。而且,目前使用的各类抗抑郁药都能够提高海马及前额叶的PI3K/AKT/mTOR通路的活性[46]。而雷帕霉素能够通过抑制mTOR抵消上述抗抑郁作用[47]。
目前,临床主要的抗抑郁药需要服用数周乃至数月后起效,因此新的药物开发主要聚焦快速起效以及对难治性抑郁的效果。快速起效的抗抑郁药如氯胺酮(Ketamine),在难治性抑郁症患者身上表现出的快速、持久、强大的疗效。研究[48]表明,氯胺酮能够通过激活前额叶AKT来提高mTOR活性,促进突触相关蛋白的合成,影响突触可塑性,从而达到治疗作用。目前,氯胺酮鼻腔内给药制剂(艾氯胺酮,esketamine)近期已在美国上市并运用于治疗难治性抑郁症,在治疗抑郁症方面有巨大潜力。
3.5 中枢神经系统肿瘤 PI3K/AKT/mTOR信号通路是各种肿瘤发生的经典信号通路,与中枢神经系统肿瘤发生也关系密切。胶质细胞瘤患者的mTOR1,2途径均异常激活[49]。恶性胶质瘤通常伴有PTEN变异和mTOR过度激活,而且基本上所有的恶性胶质瘤都有EGF(PI3K的激活分子之一)的过度表达[50]。
PI3K/AKT/mTOR信号通路抑制剂作为辅助治疗是抗肿瘤药物研发的热点方向之一。特异性mTOR抑制剂治疗胶质母细胞瘤,均有一定效果[51⁃52]。但PI3K/AKT/mTOR信号通路抑制剂只能一定程度限制肿瘤细胞生长,而且特异性不强,对比手术、放化疗等传统治疗方法,只能作为辅助治疗。寻找细胞毒性更强,特异性更好的PI3K/AKT/mTOR信号通路抑制剂将对多种神经系统肿瘤乃至其他癌症的治疗都有重要意义。
3.6 皮质发育畸形 PI3K/AKT/mTOR通路调控神经元增殖,在大脑发育过程中有重要作用。但发育过程中该通路的过度激活会导致大脑过度生长,表现为各类皮质发育畸形(malformations of cor⁃tical development,MCDs)。许多类型的MCDs均有PI3K、AKT基因变异[53]。此外,PTEN基因变异后,PI3K/AKT/mTOR通路失去有效的负调控机制,过度激活,导致细胞增殖过度,表现为巨脑症[54]。
对MCDs的诊断主要依靠影像学的形态学特征改变,但对于患病程度较轻的胎儿,诊断有一定困难,此外形态学改变往往表现在胎儿发育的中后期,而早期诊断有利于减轻父母精神和家庭负担,至关重要。对上述基因的变异进行检测鉴定,无疑为MCDs的早期诊断提供了便利。
4 总结与展望
PI3K/AKT/mTOR信号通路广泛存在于各种神经细胞中,其中PI3K分布于细胞膜周围,接受细胞外刺激,启动该通路,进而激活下游的AKT/mTOR,促进神经元存活、轴突再生、神经发生、神经元增殖分化、突触可塑性,抑制自噬。而PTEN则作为重要的负调控机制,能够防止其过度激活。AD、PD、HD属于常见的神经退行性病变,这些疾病一个重要的共同点便是神经元内病理产物的异常堆积,影响神经元正常生理活动,甚至导致神经元死亡。这些疾病的PI3K/AKT/mTOR信号通路过度激活,导致自噬受抑制,加重病理产物沉积,故抑制该通路能一定程度减轻这些疾病的病情。反之,PI3K/AKT/mTOR信号通路受抑制时,神经元的突触可塑性受损,导致抑郁症的发生。而PI3K/AKT/mTOR信号通路相关蛋白基因突变,常导致通路的异常激活,成年人表现为中枢神经系统肿瘤,胚胎则发展为皮质发育畸形。
因此,中枢神经系统PI3K/AKT/mTOR信号通路过度激活或过度抑制,都会导致病变。目前研究对PI3K/AKT/mTOR信号通路在这些脑神经疾病发生发展过程中的作用有了较为深入的解释,但一部分机制仍不明确。此外,如何人为调控PI3K/AKT/mTOR信号通路,从而治疗上述脑神经疾病也是一大挑战。应当继续中枢神经系统PI3K/AKT/mTOR信号通路的研究,有望为以上脑神经疾病的机制研究和治疗提供更多可靠的依据。
