进行性骨发育异常1例
2020-03-26胡海怡樊翌明黎兆军莫玉兰
胡海怡, 樊翌明, 黎兆军, 莫玉兰
(1.广东医科大学研究生院, 2.广东医科大学附属医院皮肤科,广东 湛江 524000)

图1进行性骨发育异常临床表现1A、1B:患儿8个月时右上肢出现暗红色丘疹、结节;1C、1D:患儿20个月时皮疹增多、增大
Fig.1Clinical pictures of the patient with progressive osseous heteroplasia.1A,1B: Dark red papules and nodules on the right back of hand and arm at8months;1C,1D: Increased number of rash at20months.
1 临床资料
患儿女,8个月11天。因“右上肢暗红色丘疹、结节7个月”于 2018年5月在我科就诊。患儿出生1月余后,父母发现其右上肢多发性暗红色丘疹、结节,此后皮疹增多、增大,影响腕关节活动。患儿系G3P3,孕38周顺产,出生体重2.1 kg,出生后无窒息,母乳喂养。父母为非近亲结婚,母孕期无异常。父母及2个兄长均无类似皮疹。
体格检查:发育正常,正常面容。右上臂屈侧、肘窝、前臂屈侧、腕部、手背可见多发性暗红色谷粒至绿豆大小丘疹、结节、斑块,边界清楚,质较硬,可活动,与周围组织无明显黏连(图1A、1B)。血常规及血清甲状腺素、甲状旁腺激素、钙、磷无异常。右上臂皮损组织病理检查:真皮浅层小血管扩张,血管周围淋巴组织浸润,真皮中部见散在团状或片状嗜伊红骨组织,骨陷窝内有嗜碱性骨细胞,边缘见卵圆形或长梭形的成骨细胞,骨组织中央小血管增生,周围见纤维组织形成的包膜(图2A)。茜素红钙染色显示真皮内钙质沉积(图2B)。右手正、斜位X光:右腕-手掌桡侧软组织多发结节,呈斑片状高密度影、钙化灶(图3A、3B)。

图2右上臂皮损组织病理检查2A:真皮浅层小血管扩张,血管周围淋巴组织浸润,真皮中部见散在团状或片状嗜伊红骨组织,骨陷窝内有嗜碱性骨细胞,边缘见卵圆形或长梭形的成骨细胞,骨组织中央小血管增生,周围见纤维组织形成的包膜(HE,100×);2B:钙质沉积(茜素红钙染色,100×)
Fig.2Histopathology of right upper arm.2A: Dilation of small blood vessels and perivascular infiltration of lymphocytes in the upper dermis.Focal and scattered bone tissue with basophilic osteocytes surrounded by oval or long spindle-shaped osteocytes in the middle dermis.Bone tissues with hyperplasia of small vessels were wrapped by fibrous tissues.Oval and long spindle-shaped osteocytes.(HE,100×);2B: Calcium deposition (Alizarin red calcium staining,100×)
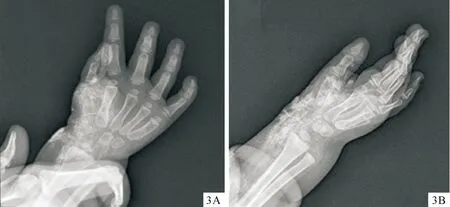
图3右手正位(3A)、斜位(3B)X光检查:右手腕部-手掌桡侧软组织多发性高密度钙化灶
Fig.3X-ray examination of the right hand in positive (3A) and oblique (3B) positions: multiple hyperdense calcification of soft tissue at the right wrist-radial side of the palm.
GNAS基因检测:患儿及父母外周血送至深圳华大临床检验中心,采用二代测序法检测基因突变。结果显示:患儿及父亲GNAS基因5号外显子c.417delC突变(编码区第417号核苷酸胞嘧啶缺失),母亲未见异常(图4)。
诊断:进行性骨发育异常。患儿未接受任何治疗,20个月时复诊发现右上肢皮疹增多、增大,以右腕关节处明显(图1C、1D),腕关节活动受限加重。
2 讨论
进行性骨发育异常(progressive osseous heteroplasia,POH)是一种致残性异位骨化性疾病,由Kaplan等[1]于1994年首次报道。POH多为散发性,少数为家族性,表现为出生时或婴儿期发生皮肤、皮下组织异位骨化,儿童期进行性发展,女性多于男性[2-3]。1998年Urtizberea等[4]首次证明该病为常染色体显性遗传。2000年Kaplan等[5]再次发现本病存在鸟嘌呤核苷酸结合蛋白α刺激活性肽(guanine nucleotide-binding protein, α-stimulating acitivity polypeptide,GNAS)基因失活突变。 GNAS(NM_000516.4)基因定位于20q13.32,含有13个外显子和12个内含子,主要编码Gs蛋白α亚基(Gsα)[6]。Gs蛋白由α、β、γ亚基构成,在细胞间联系中起重要作用,其中α亚基是活性亚基,具有GTP、GDP结合位点。当Gsα蛋白被其表面的偶联受体激活时,α亚基从β、γ亚基复合物中解离出来,与GTP结合呈活性状态,活化的α亚基激活腺苷酸环化酶(AC),产生环磷腺苷(cAMP)。GNAS基因失活突变,减少cAMP形成,导致异位骨化形成[7]。Gsα活化可调控多种细胞代谢过程,包括成骨、脂肪、软组织形成等,维持间充质干细胞成脂与成骨的平衡[8]。GNAS基因和cAMP信号在早期、晚期胚胎发育的成骨分化中有不同作用:在胚胎发育早期,上调cAMP抑制成骨细胞分化、促进脂肪形成,而下调cAMP促进成骨细胞分化;在胚胎发育晚期,上调cAMP对矿化和骨钙素表达无影响;提示cAMP调控早期成骨细胞发生[9]。cAMP增加可抑制成骨标志物表达,促进脂肪生成标志物表达[2]。Hedgehog信号抑制脂肪形成、促进成骨分化。Gsα在Wnt/β-catenin和Hedgehog信号通路介导异位骨化过程中起关键作用,Gsα获得功能性突变激活Wnt/β-catenin信号,与进行性骨化性纤维发育不良(fibrodysplasia ossificians progressive, FOP)有关[10];而Gsα功能缺失突变激活Hedgehog信号,与POH、Albright遗传性骨营养不良(Albright hereditary osteodystrophy, AHO)相关[11]。

图4GNAS基因5号外显子测序图:患儿及父亲GNAS基因5号外显子c.417delC突变,母亲未见异常
Fig.4Sequence diagram of GNAS gene exon5: the child and her father GNAS gene showed exon5c.417delC mutation, her mother showed no abnormality.
本例患者及其父亲GNAS基因5号外显子发生c.417delC 突变,导致编码第140号氨基酸(苯丙氨酸)的密码子变为亮氨酸的密码子 (p.Phe140Leu),至今没有该变异致病性的相关报道。目前发现7号外显子突变最多[6, 12]。该患儿突变位点与父亲相同,但父亲无临床表现。Shore等[2]在2002年曾报道有GNAS基因突变不外显的案例,但同样一些患POH的患者却未出现基因失活突变(约30%的病例)。GNAS基因并不是POH的特有致病基因,除了与其他原发性骨化性疾病相关,还与多囊卵巢综合征、偏头痛、一些恶性肿瘤(肺癌等)相关[13],存在GNAS基因突变者也可无任何皮肤改变。以上可以解释为什么患儿及其父亲都存在GNAS基因突变,而其父亲无类似皮疹。
Pignolo等[8]提出了POH诊断标准:①主要指标:浅表和深部异位骨化,Albright遗传性骨营养发育不良的两个或更少的特征,无甲状旁腺激素抵抗;②次要指标:GNAS基因突变,父系遗传,网状骨化结构,包括膜内骨化或同时具有膜内和软骨内骨化的X线影像证据,宫内发育迟缓,消瘦,发病年龄<1岁。Albright遗传性骨营养发育不良的特点[14]:身材矮小、中心型肥胖、短颈、圆脸、第四和第五掌(趾)骨粗短、畸形、智力低下等。
与GNAS基因相关的异位骨化性疾病包括AHO、假性甲状旁腺功能减退症、FOP、皮肤板层状骨瘤。FOP临床病理特点是先天性双足大拇趾畸形,拇外翻,结缔组织进行性骨化[14]。POH与皮肤板层状骨瘤具有相似的临床病理特点,主要区别是POH的异位骨化呈进行性发展[15]。
本例患儿出生后1个月患病,为新生儿期发病,智力正常,右上肢皮肤开始表现为质硬的斑丘疹、丘疹、结节,1年后随访,病情缓慢发展累及深部结缔组织,右上肢斑丘疹、丘疹、结节增多并增大,右手腕关节、肘窝处活动明显受限。结合皮肤病理检查、影像学检查、基因检测,诊断POH明确。目前缺乏有效治疗。局限性异位骨化可行手术切除,而弥漫性者术后复发率较高。GNAS突变类型、定位不能预测POH进展程度[1]。皮肤钙化可引起生长发育迟缓、肢体不等长、关节活动障碍、皮肤溃疡、继发性感染等[16]。这主要取决于异位骨化的程度和深度[2, 6]。
