实验性牙周炎大鼠脑内外的炎性改变
2020-03-19宋忠臣
胡 苡 ,周 薇,宋忠臣
1.上海交通大学医学院附属第九人民医院·口腔医学院牙周病科,国家口腔疾病临床医学研究中心,上海市口腔医学重点实验室,上海市口腔 医学研究所,上海 200011;2. 上海交通大学医学院附属第九人民医院· 口腔医学院口腔微生态与系统性疾病实验室,国家口腔疾病临床医学研究中心,上海市口腔医学重点实验室,上海市口腔医学研究所,上海 200125
牙周炎(periodontitis)是一种微生物相关的、宿主介导的、多因素参与并导致牙周附着丧失的炎症性疾病[1-2]。牙龈卟啉单胞菌(Porphyromonas gingivalis,P. gingivalis) 是牙周炎主要致病菌之一,脂多糖 (lipopolysaccharide,LPS) 是其主要毒力因子,有促进炎症激活和放大的作用。牙周局部炎症会破坏上皮的完整性,使牙周袋内的炎症介质如白细胞介素(interleukin,IL)等细胞因子从牙周袋进入血液循环系统和外周免疫器官,引起系统性炎症反应[3]。因此,牙周慢性反复感染和宿主炎症免疫反应不仅导致牙周支持组织的破坏,也成为许多系统性疾病如心血管疾病、糖尿病、呼吸系统疾病等的重要危险因素[4]。近年来探讨牙周炎和系统性疾病的共同危险因素及可能机制已成为研究热点。
牙周炎患者外周循环中促炎因子的表达量明显高于健康人群,提示炎症反应及其释放的炎症因子可能是牙周炎与多种系统性疾病关联的机制[5]。牙周炎可使全身处于微炎症状态,诱发神经炎症反应,通过炎症机制促进包括阿尔茨海默病(Alzheimer's disease,AD)在内的系统性疾病的发生和发展[6-7]。AD 是一种以进行性认知功能障碍为主要临床症状的神经退行性疾病,已成为继心脑血管疾病、恶性肿瘤之后威胁老年人生命的第三大疾病[8]。脑内炎症是AD 的关键病理标志之一,认知状态恶化与促炎介质入脑有关[9-10]。在AD 患者的血清和脑脊液中检测到IL-1β、IL-6 等牙周炎相关细胞因子表达上调,与认知功能衰退、AD 风险进一步升高有关[11-13]。但目前牙周炎与AD的相关机制尚不清楚。
流行病学调查提示牙周炎与系统性疾病密切相关[4]。本课题组前期研究已证实,急性腹腔注射P. gingivalis-LPS 可诱导神经炎症[14]。但实验性牙周炎对外周及脑内炎症状态的影响尚不清楚。因此,本研究拟观察龈沟注射P. gingivalis-LPS 建立的实验性牙周炎对大鼠外周血、外周免疫器官及中枢内炎症水平的影响,为证实牙周局部感染能够诱发外周炎症及脑内炎症提供实验依据,以进一步探讨牙周炎在AD 进程中的可能作用。
1 材料与方法
1.1 实验材料与仪器
1.1.1 实验动物 8 ~10 周龄健康雄性SD 大鼠,体质量250 ~300 g,由上海交通大学医学院附属第九人民医院中心实验室[实验动物使用许可证号SYXK(沪)2016-0016]提供,饲养在控制光、温度和湿度的SPF 级环境中,并允许其自由获得食物和水。本研究经上海交通大学医学院附属第九人民医院动物伦理委员会批准(批件号SH9H-2019-A465-1)。
1.1.2 主要试剂与仪器 P. gingivalis-LPS、TAK-242(Invivogen,美 国),RNAiso Plus、PrimeScript®反 转 录 试 剂 盒(Takara,日本),LightCycler®480(Roche,瑞士),Iba1(Arigo Biolaboratories,中国台湾),GFAP(Abcam,美国),激光共聚焦显微镜(Leica TCS SP2,德国)。
1.2 龈沟内注射P. gingivalis-LPS 建立大鼠实验性牙周炎模型及实验分组
8 ~10 周 龄SD 大 鼠 随 机 分4 组(n=6),具 体 分组情况如下。①LPS 组:双侧上颌第一磨牙龈沟内注射P. gingivalis-LPS。 ②LPS+TAK-242 组: 腹 腔 注 射TAK-242,1 h 后向双侧上颌第一磨牙龈沟内注射等剂量的P. gingivalis-LPS。③TAK-242 组:腹腔注射TAK-242。④ 对照组(control 组):腹腔注射等量生理盐水。
采用腹腔注射10% 水合氯醛(0.4 mL/100 g)麻醉大鼠。麻醉满意后,对每只大鼠进行龈沟内注射 (P. gingivalis-LPS)、腹腔内注射(TAK-242),药物剂量均为每次0.5 mg/kg,每周2 次,持续10 周。
1.3 苏木精-伊红染色
苏 木 精- 伊 红 染 色(hematoxylin-eosin staining,H-E staining)方法如下。按照每 100 g 体质量,腹腔注射0.4 mL的10%水合氯醛麻醉大鼠。麻醉后取大鼠上颌骨,置于4%多聚甲醛中固定,4 ℃过夜。将上颌骨置于10%的EDTA脱钙液中,每3 ~5 d 换液1 次,持续4 ~6 周,直至针能无阻力穿透上颌骨。切取大鼠上颌第一磨牙段组织块,常规石蜡包埋,切片,脱蜡,苏木素染色,冲洗,分化,浸泡,伊红染色,冲洗,浸泡,脱水,透明,封片。
1.4 免疫组织化学染色观察脑内小胶质细胞及星形胶质细胞
腹腔注射10%水合氯醛(0.4 mL/100 g)麻醉大鼠。麻醉满意后取材,固定、脱水、透明、浸蜡、包埋、切片、粘片、烤片、染色。采用离子钙结合蛋白1(ionized calcium binding adapter molecule 1,Iba1)标记脑内的小胶质细胞,胶质纤维酸性蛋白(glial fibrillary acidic protein,GFAP)标记脑内的星形胶质细胞,采用LEICA 显微镜对每张切片进行拍摄。
1.5 实时定量荧光检测
用实时定量荧光 PCR(real-time PCR,RT-PCR)检测大鼠外周及中枢内IL-1β、IL-6、IL-8 基因的表达水平。
大鼠麻醉后采集腹主动脉血,每只大鼠采集4 ~5 mL血,置于肝素钠抗凝采血管内。无菌条件下用等体积灭菌PBS 稀释抗凝管内剩余的3 ~3.5 mL 血,混匀待用。使用Ficoll 对外周血进行密度梯度离心(800×g,25 min),分离得到外周血单核淋巴细胞(peripheral blood mononuclear cell,PBMC)。研磨大鼠脾脏及脑组织得到组织匀浆。
采用TRIzol 法提取RNA,按PrimeScript®反转录试剂盒说明书进行反转录。得到cDNA 后按SYBR®Primix Ex Taq ™ Ⅱ说明书,采用磷酸甘油醛脱氢酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)作为内参基因,采用Light Cycler®480 进行RT-PCR。RT-PCR 反应条件及流程:预变性(98 ℃,5 min),变性(98 ℃,15 s,40 循环),退火(55 ℃,15 s),延伸(72 ℃,30 s)。引物序列见表1。
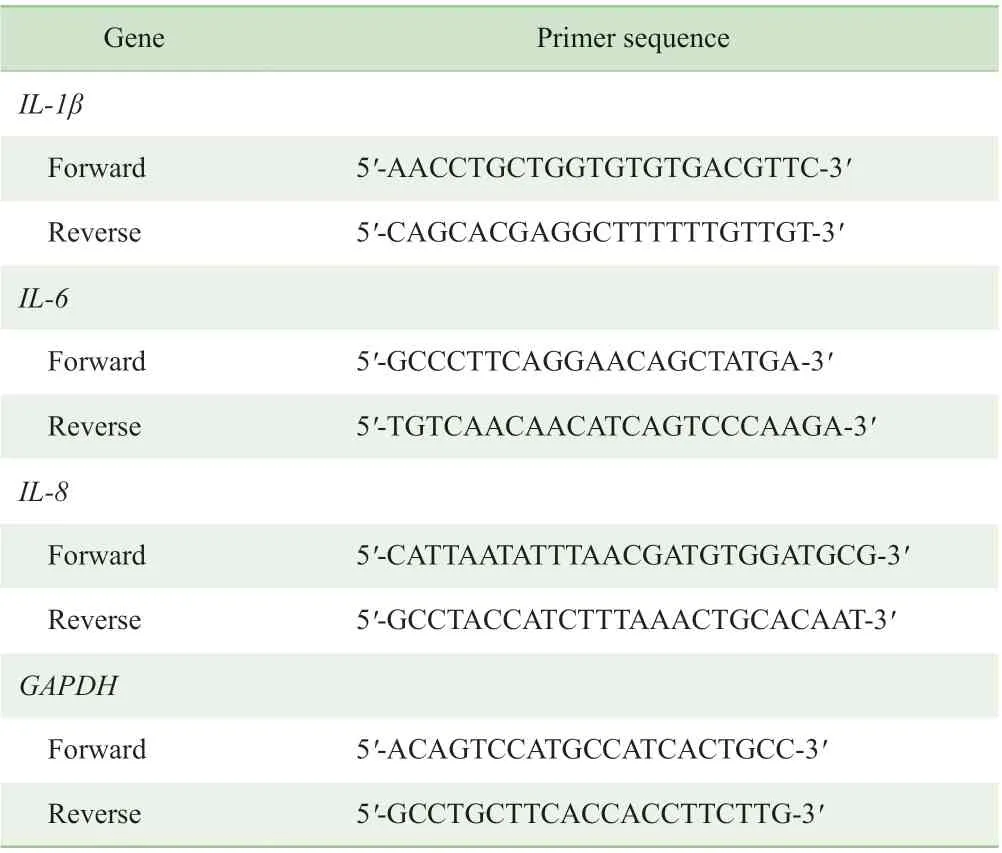
表1 基因引物序列Tab 1 Primer sequences of target genes
1.6 统计学方法
采用Graphd Prism5.0 软件进行统计分析,定量数据以±s 表示,组间比较采用单因素方差分析,进一步采用Tukey 法进行2 组间的比较。P<0.05 表示差异有统计学意义。
2 结果
2.1 实验性牙周炎大鼠的牙周组织病理变化
通过H-E 染色观察大鼠上颌第一磨牙区的牙周组织状况。对照组与TAK-242 组大鼠牙槽骨未见明显吸收,骨表面较平整光滑,纤维排列整齐清晰;而龈沟注射P. gingivalis-LPS 可造成大鼠上颌第一磨牙牙槽骨高度下降,与对照组相比,牙槽骨高度吸收至根尖1/3;且牙槽骨表面出现不规则吸收凹陷,伴大量炎症细胞浸润,纤维排列紊乱。在LPS+TAK-242 组,第一磨牙区的牙槽骨水平下降及炎症反应得到部分逆转(图1)。H-E 染色结果表明,龈沟注射P. gingivalis-LPS 可导致牙周炎样病理改变,实验性牙周炎模型成功建立。
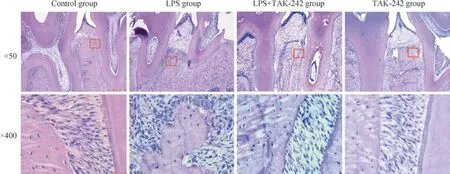
图1 龈沟注射P. gingivalis-LPS 诱导的牙周炎对大鼠牙周组织的影响(H-E 染色)Fig 1 Effects of periodontitis induced by P. gingivalis-LPS on periodontal tissues in rats (H-E staining)
2.2 实验性牙周炎大鼠PBMCs 中促炎因子表达水平的变化
如图2 所示,对照组与TAK-242 组大鼠PBMCs 的mRNA 表达水平均无统计学差异。与对照组相比,龈沟注射P. gingivalis-LPS 可显著上调大鼠PBMCs 中IL-1β(P=0.027)、IL-6(P=0.007)、IL-8(P=0.038) 的mRNA表达。LPS+TAK-242 组与LPS 组相比,上述因子的上调均受到明显抑制(P=0.010,P=0.013,P=0.020)。结果显示,龈沟注射P. gingivalis-LPS 诱导的实验性牙周炎可上调大鼠PBMCs 中促炎因子如IL-1β、IL-6、IL-8 的表达水平,且上述变化可部分被TAK-242 阻断。
2.3 实验性牙周炎大鼠脾脏中促炎因子表达水平的变化
如图3 所示,对照组与TAK-242 组大鼠脾脏的mRNA 表达水平均无统计学差异。与对照组相比,龈沟注射P. gingivalis-LPS 可显著上调大鼠脾脏中IL-6(P=0.033)和IL-8(P=0.001) 的mRNA 表 达。LPS+TAK-242 组 与LPS 组相比,IL-1β(P=0.027)、IL-8(P=0.005)的上调均受到明显抑制;IL-6 的表达呈下调趋势,但差异无统计学意义。结果显示,龈沟注射P. gingivalis-LPS 诱导的实验性牙周炎可上调大鼠脾脏中促炎因子IL-1β、IL-6、IL-8的表达水平,且上述变化可部分被TAK-242 阻断。

图2 龈沟注射P. gingivalis-LPS 诱导的牙周炎对大鼠PBMCs 中促炎因子mRNA 表达水平的影响Fig 2 Effects of periodontitis induced by P. gingivalis-LPS on the mRNA expression of proinflammatory factors in the PBMCs of rats
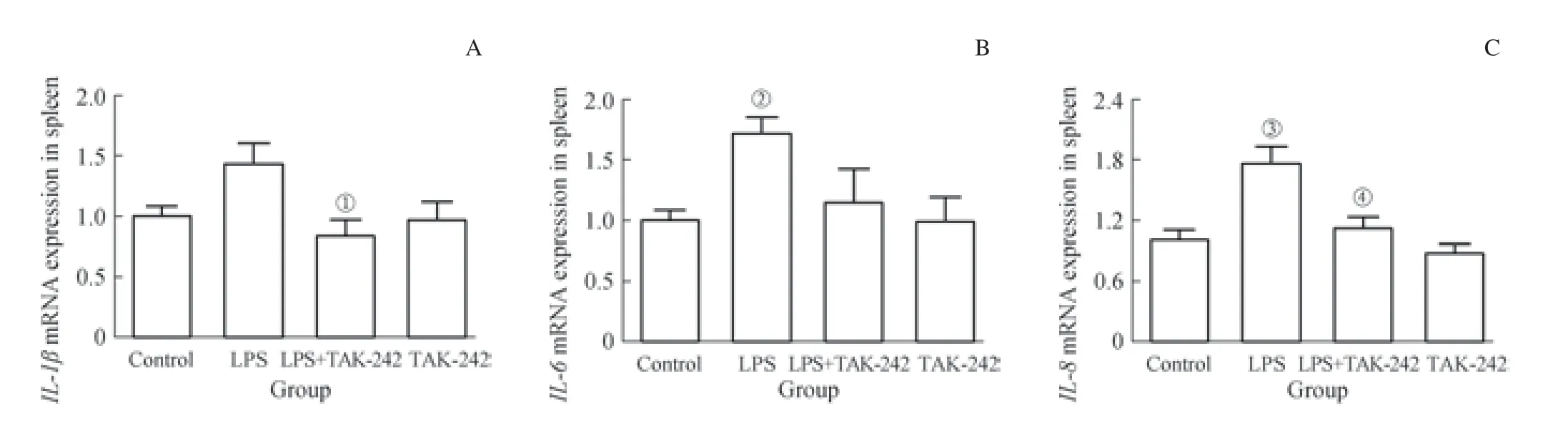
图3 龈沟注射P. gingivalis-LPS 诱导的牙周炎对大鼠脾脏中促炎因子mRNA 表达水平的影响Fig 3 Effects of periodontitis induced by P. gingivalis-LPS on the mRNA expression of proinflammatory factors in the spleen of rats
2.4 实验性牙周炎大鼠脑内海马中小胶质细胞及星形胶质细胞的变化
在大鼠脑内海马中可见到小胶质细胞被Iba1 染色。在对照组、LPS+TAK-242 组和TAK-242 组中仅有少量活化的小胶质细胞;而在LPS 组中可观察到具有不规则突起的、活化的小胶质细胞数量明显增加(图4A)。说明龈沟注射P. gingivalis-LPS 诱导的牙周炎可激活海马的小胶质细胞,且上述激活作用可被TAK-242 部分阻断。
在大鼠脑内海马中可见到星形胶质细胞被GFAP 染色。在对照组,LPS+TAK-242 组和TAK-242 组中仅有少量活化的星形胶质细胞;而在LPS 组中可观察到胞体增大、不规则突起增加的星形胶质细胞数量明显增加(图4B)。说明龈沟注射P. gingivalis-LPS 诱导的实验性牙周炎可激活海马的星形胶质细胞,且上述激活作用可被TAK-242 部分阻断。
2.5 实验性牙周炎大鼠海马内促炎因子表达水平的变化
如图5 所示,对照组与TAK-242 组大鼠内海马中各促炎因子的mRNA 表达水平均无统计学差异。与对照组相比,龈沟注射P. gingivalis-LPS 可显著上调大鼠脑组织中IL-1β(P=0.040)、IL-6(P=0.035)、IL-8(P=0.010)的mRNA 表达。LPS+TAK-242 组与LPS 组相比,IL-1β(P=0.018)、IL-6(P=0.042)的上调均受到明显抑制;IL-8 的表达呈下调趋势,但差异无统计学意义。结果显示,龈沟注射P. gingivalis-LPS 诱导的实验性牙周炎可上调大鼠脑组织中促炎因子IL-1β、IL-6、IL-8 的表达水平,且上述变化可部分被TAK-242 阻断。
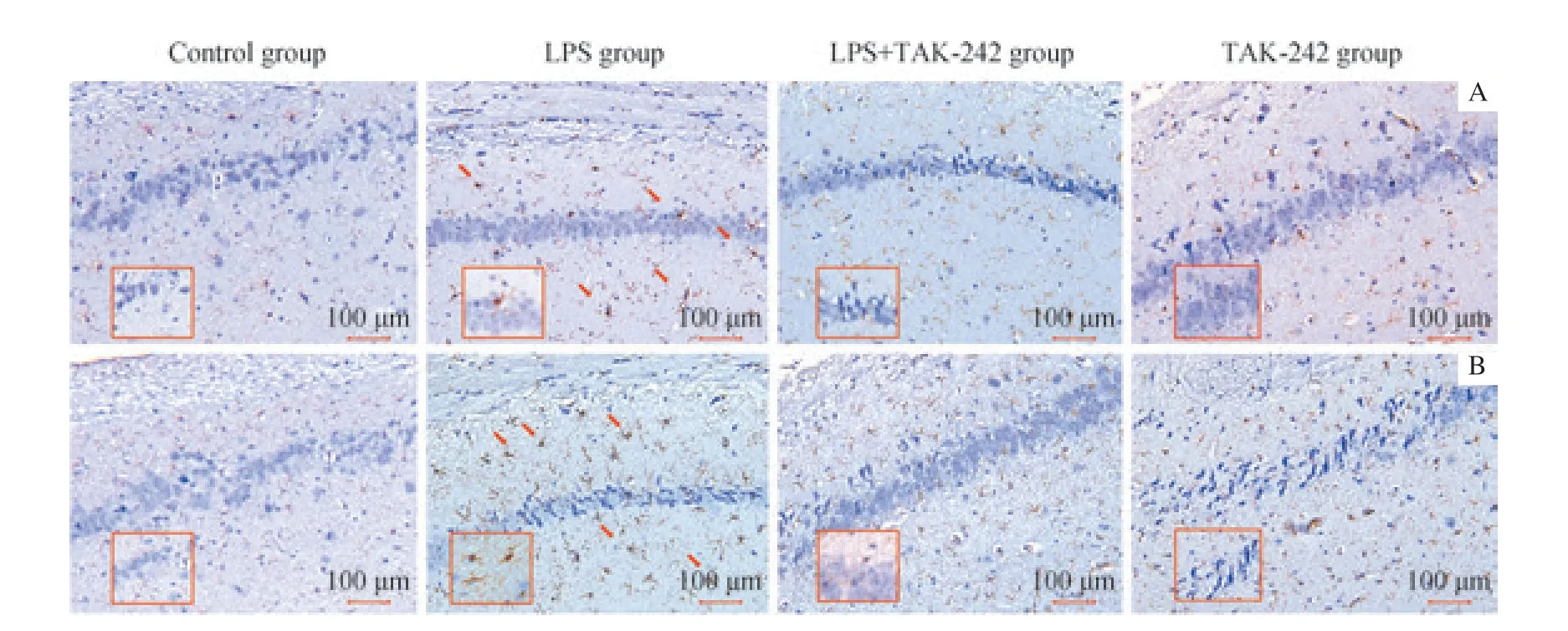
图4 龈沟注射P. gingivalis-LPS 诱导的牙周炎对大鼠海马内小胶质细胞及星形胶质细胞的影响(DAB 染色,×200)Fig 4 Effects of periodontitis induced by P. gingivalis-LPS on microglia and astrocytes in the hippocampus of rats (DAB staining, ×200)
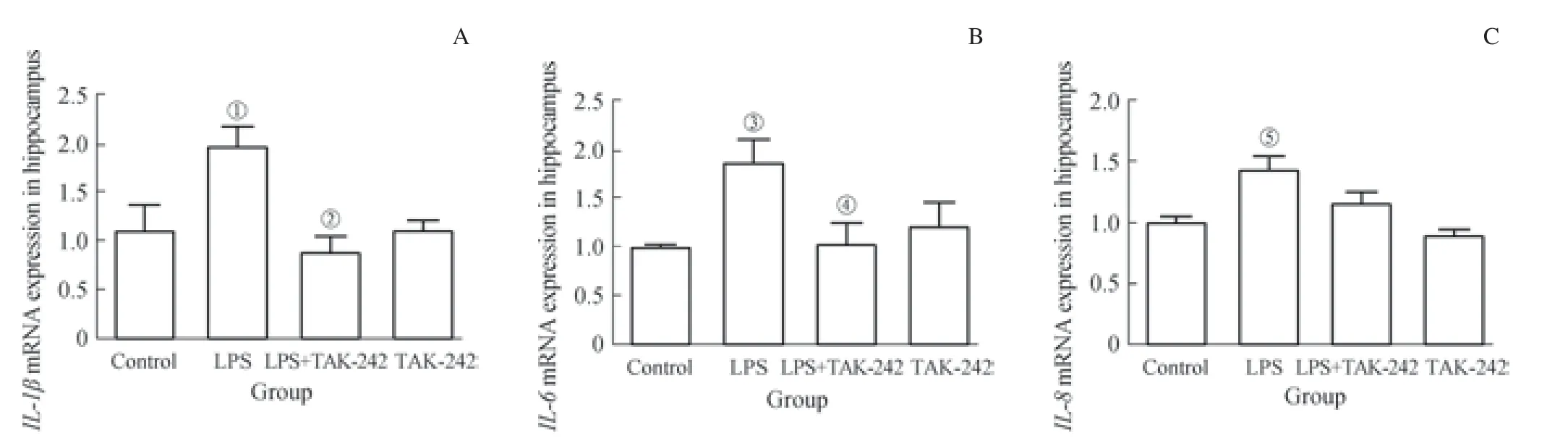
图5 龈沟注射P. gingivalis-LPS 诱导的牙周炎对大鼠海马中促炎因子mRNA 表达水平的影响Fig 5 Effects of periodontitis induced by P. gingivalis-LPS on the mRNA expression of proinflammatory factors in the hippocampus of rats
3 讨论
P. gingivalis-LPS 不仅可以破坏牙周支持组织,同时可造成全身微炎症状态,扩散至中枢神经系统[15]。目前尚未有研究报道龈沟注射P. gingivalis-LPS 建立的实验性牙周炎对外周及神经炎症的影响 。
本研究参考Li 等[16]的实验方法,采用向大鼠双侧上颌第一磨牙龈沟内注射P. gingivalis-LPS 的方法建立实验性牙周炎模型。H-E 染色结果证实LPS 组大鼠的牙槽骨明显吸收,骨表面出现不规则凹坑状骨吸收,伴有炎症细胞浸润、纤维排列紊乱等牙周炎样病理变化,提示牙周炎模型建立成功。
牙周炎释放的免疫炎症因子进入系统性循环可诱发系统性炎症。牙周炎可上调牙周组织内促炎因子IL-1β、IL-6的水平,上述因子进入系统性循环后可对远隔组织及器官产生影响[17-18]。健康人群的外周循环中促炎因子表达水平极低,然而在牙周炎患者体内,除牙周组织局部促炎因子水平升高外,外周循环中也可检测到促炎因子表达明显增加[5]。脾脏作为机体重要的淋巴器官和第二大免疫器官,在调节炎症反应中发挥重要作用[19]。有研究[19]发现E. coli-LPS能够诱发脾组织释放大量促炎因子进入体循环。本实验发现,龈沟注射P. gingivalis-LPS 建立的实验性牙周炎可导致大鼠外周血及脾脏中促炎因子IL-1β、IL-6、IL-8 的表达明显上调,证实牙周炎可引发外周血及外周免疫器官炎症。
神经炎症主要是由小胶质细胞和星形胶质细胞的活化以及细胞因子、趋化因子或生长因子的释放引起的,且神经炎症通常发生在AD 初期[20]。Venneti 等[21]已证实,活化的小胶质细胞可分泌促炎介质,过度活化的星形胶质细胞也可导致炎症细胞因子的产生,并导致神经元死亡[22]。脑组织中普遍存在炎症反应,小胶质细胞和星型胶质细胞呈簇状集中于淀粉样斑块周围,神经血管单元的“炎症网络”加剧神经元丢失,在AD 形成和发展过程中具有关键性作用。本实验发现,LPS 组大鼠脑组织内由Iba1 标记的小胶质细胞和GFAP 标记的星形胶质细胞均呈激活状态,给予TAK-242 可阻断上述变化。
持续性的外周和中枢炎症及其连锁反应是AD 炎症病理的关键特征[23]。腹腔注射P. gingivalis-LPS 可诱导神经炎症[11]。IL-1β 的水平在AD 患者脑中持续上调,是AD的关键分子之一;已证实IL-1β 可促进活化的小胶质细胞释放多种炎症介质,进而激活星形胶质细胞,形成细胞因子“正反馈”导致神经炎症[24-26]。IL-6 可介导AD 患者脑内免疫反应,参与神经炎症和老年斑的形成过程,在AD患者的血浆、脑脊液、脑内斑块中表达明显增加,且其表达水平随着AD 进展上调,可导致神经细胞损伤[27-28]。IL-8 作为趋化因子可发挥炎症趋化作用,在AD 状态下吸引中性粒细胞至脑内病变区加重损伤,在AD 患者的血清和脑脊液内浓度均明显高于健康对照组[29-30]。本实验证实龈沟注射P. gingivalis-LPS 建立的实验性牙周炎可上调大鼠脑组织内IL-1β、IL-6、IL-8 的表达水平,促进神经炎症发展。
本实验发现龈沟注射P. gingivalis-LPS 建立的实验性牙周炎模型可以通过引发大鼠外周血和以脾为代表的外周免疫器官炎症,同时使脑内小胶质细胞、星形胶质细胞活化,促进脑内神经炎症;即牙周炎可通过外周及中枢的炎症反应促进AD 的发生和发展。
参·考·文·献
[1] Papapanou PN, Sanz M, Buduneli N, et al. Periodontitis: consensus report of workgroup 2 of the 2017 world workshop on the classification of periodontal and peri-implant diseases and conditions[J]. J Periodontol, 2018, 89(Suppl 1): S173-S182.
[2] 孟焕新.2018 年牙周病和植体周病国际新分类简介[J].中华口腔医学杂志, 2019, 54(2): 73-78.
[3] Deo V, Bhongade ML. Pathogenesis of periodontitis: role of cytokines in host response[J]. Dent Today, 2010, 29(9): 60-62, 64-6; quiz 68-69.
[4] Kumar PS. From focal Sepsis to periodontal medicine: a century of exploring the role of the oral microbiome in systemic disease[J]. J Physiol, 2017, 595(2): 465-476.
[5] Loos BG. Systemic markers of inflammation in periodontitis[J]. J Periodontol, 2005, 76(11 Suppl): 2106-2115.
[6] Singhrao SK, Chukkapalli S, Poole S, et al. Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE-/-mice brains[J]. J Oral Microbiol, 2017, 9(1): 1270602.
[7] Halliday MR, Rege SV, Ma QY, et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease[J]. J Cereb Blood Flow Metab, 2016, 36(1): 216-227.
[8] Wimo A, Guerchet M, Ali GC, et al. The worldwide costs of dementia 2015 and comparisons with 2010[J]. Alzheimers Dement, 2017, 13(1): 1-7.
[9] Maccioni RB, González A, Andrade V, et al. Alzheimer's disease in the perspective of neuroimmunology[J]. Open Neurol J, 2018, 12: 50-56.
[10] Haddick PC, Larson JL, Rathore N, et al. A common variant of IL-6R is associated with elevated IL-6 pathway activity in Alzheimer's disease brains[J]. J Alzheimers Dis, 2017, 56(3): 1037-1054.
[11] Kaye EK, Valencia A, Baba N, et al. Tooth loss and periodontal disease predict poor cognitive function in older men[J]. J Am Geriatr Soc, 2010, 58(4): 713-718.
[12] Kim YS, Lee KJ, Kim H. Serum tumour necrosis factor-α and interleukin-6 levels in Alzheimer's disease and mild cognitive impairment[J]. Psychogeriatrics, 2017, 17(4): 224-230.
[13] Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: current evidence and future directions[J]. Alzheimers Dement, 2016, 12(6): 719-732.
[14] Zhang J, Yu CB, Zhang X, et al. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice[J]. J Neuroinflammation, 2018, 15(1): 37.
[15] Dominy SS, Lynch C, Ermini F, et al. Porphyromonas gingivalis in Alzheimer's disease brains: evidence for disease causation and treatment with smallmolecule inhibitors[J]. Sci Adv, 2019, 5(1): eaau3333.
[16] Li XT, Yu CB, Hu Y, et al. New application of psoralen and angelicin on periodontitis with anti-bacterial, anti-inflammatory, and osteogenesis effects[J]. Front Cell Infect Microbiol, 2018, 8: 178.
[17] Ng A, Tam WW, Zhang MW, et al. IL-1β, IL-6, TNF- α and CRP in elderly patients with depression or Alzheimer's disease: systematic review and metaanalysis[J]. Sci Rep, 2018, 8: 12050.
[18] Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity[J]. Immunity, 2013, 39(5): 806-818.
[19] Semaeva E, Tenstad O, Skavland J, et al. Access to the spleen microenvironment through lymph shows local cytokine production, increased cell flux, and altered signaling of immune cells during lipopolysaccharide-induced acute inflammation[J]. J Immunol, 2010, 184(8): 4547-4556.
[20] Michelucci A, Heurtaux T, Grandbarbe L, et al. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-β[J]. J Neuroimmunol, 2009, 210(1-2): 3-12.
[21] Venneti S, Wang GJ, Nguyen J, et al. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders[J]. J Neuropathol Exp Neurol, 2008, 67(10): 1001-1010.
[22] Choi SS, Lee SR, Lee HJ. Neurorestorative role of stem cells in Alzheimer's disease: astrocyte involvement[J]. Curr Alzheimer Res, 2016, 13(4): 419-427.
[23] Lyman M, Lloyd DG, Ji XM, et al. Neuroinflammation: the role and consequences[J]. Neurosci Res, 2014, 79: 1-12.
[24] Lai KSP, Liu CS, Rau A, et al. Peripheral inflammatory markers in Alzheimer's disease: a systematic review and meta-analysis of 175 studies[J]. J Neurol Neurosurg Psychiatry, 2017, 88(10): 876-882.
[25] Mendiola AS, Cardona AE. The IL-1β phenomena in neuroinflammatory diseases[J]. J Neural Transm (Vienna), 2018, 125(5): 781-795.
[26] Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer's disease[J]. J Cell Biol, 2018, 217(2): 459-472.
[27] Qiu ZH, Gruol DL. Interleukin-6, β-amyloid peptide and NMDA interactions in rat cortical neurons[J]. J Neuroimmunol, 2003, 139(1-2): 51-57.
[28] Bolós M, Perea JR, Avila J. Alzheimer's disease as an inflammatory disease[J]. Biomol Concepts, 2017, 8(1): 37-43.
[29] Alsadany MA, Shehata HH, Mohamad MI, et al. Histone deacetylases enzyme, copper, and IL-8 levels in patients with Alzheimer's disease[J]. Am J Alzheimers Dis Other Demen, 2013, 28(1): 54-61.
[30] Na KS, Jung HY, Kim YK. The role of pro-inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia[J]. Prog Neuropsychopharmacol Biol Psychiatry, 2014, 48: 277-286.
