气相色谱法测定水产品中丙泊酚麻醉剂的残留量
2020-03-13颜珲璘黄和高平陈日檬曾丹丹周凯
颜珲璘 ,黄和 *,高平,陈日檬,曾丹丹,周凯
1. 广东海洋大学食品科技学院(湛江 524088);2. 广东省水产品加工与安全重点实验室(湛江 524088);3. 广东普通高等学校水产品深加工重点实验室(湛江 524088);4. 湛江市食品药品检验所(湛江 524022)
随着生活物质水平的提高,人们对鲜活水产品的需求越来越多,为了降低水产品在运输过程中的损耗,渔用麻醉剂的使用开始广泛,但麻醉剂的残留量及其安全性问题也备受关注[1]。目前常使用的麻醉剂有CO2、MS-222、丁香酚类化合物等[2]。
丙泊酚化学名为2, 6-双异丙基苯酚[3],该品为高脂溶性的烷基酚类的短效静脉麻醉剂,它具有麻醉诱导起效快、苏醒迅速且功能恢复完善等优点[4],常见于医学临床使用。有研究表明,丙泊酚易引起心脏和呼吸抑制[5]。丙泊酚具有效迅速、镇静作用强的特点,其引起滥用、意外过量和自杀等案件逐年增多,特别是美国著名歌星迈克尔·杰克逊之死案件[6],更使丙泊酚受到极大关注。
目前,关于样品中丙泊酚的分析方法有高效液相法[7]、气相色谱质谱联用法[8]、液相色谱质谱联用法[9]等。丙泊酚麻醉剂的残留量检测分析主要在血液、药品等范围,有关水产品中丙泊酚麻醉剂残留量的检测方法未见报道。试验采用分散固相萃取的样品前处理方法,对水产品中的丙泊酚麻醉剂残留量的测定方法进行研究,旨在建立一种能够测定水产品中丙泊酚麻醉剂的简单、快速、灵敏的方法,为我国水产品中丙泊酚麻醉剂残留检测提供技术支撑。
1 材料与方法
1.1 材料与试剂
南美白对虾、鳜、金鲳,洗净,去鳞、去皮,取出肌肉部分,用搅拌机搅碎,放入-18 ℃冰箱中保存,备用。
丙泊酚(CAS号2078-54-8,浓度≥99%)、内标物百里香酚(CAS号89-83-8,浓度≥99%)、无水硫酸镁(上海麦克林公司);乙腈、甲醇、乙酸乙酯(色谱纯,Thermo Fisher公司);无水硫酸镁(国药集团公司);QuEchERs dSPE EMR-Lipid、EMR-polish(安捷伦公司);ProElut PLS(150 mg/6 mL,迪马科技);C18反相固相萃取柱(500 mg/3 mL,Thermos公司)。
1.2 仪器与设备
GC-2010 PLUS气相色谱仪(日本岛津公司);ST-16R型高速冷冻离心机(Thermos公司);TP-220 A型电子天平(湘仪天平仪器有限公司);VTX-3000 L型旋涡混匀器(东京理化有限公司);KQ-250 DE型超声波清洗器(昆山超声仪器有限公司);超纯水机(Thermos公司)。
1.3 方法
1.3.1 样品前处理
称取2.0 g(精确至0.01 g)1.1所述样品于50 mL离心管中,加入一粒均质子,加入10 mL乙酸乙酯,漩涡振荡1 min,离心机以5 000 r/min离心5 min,取出上清液于15 mL具塞离心管中,待净化。
1.3.2 净化
取EMR-Liplid dSPE分散净化管,加入5 mL水,摇匀,然后吸取5 mL上述待净化液至净化管中,漩涡振荡1 min,离心机以5 000 r/min离心3 min,将离心后的提取液移取5 mL至EMR Lipid Polish反萃取管中,漩涡混合,吸取1 mL净化液于进样小瓶中,待GC分析。
1.4 色谱条件
色谱柱:HP-50+(30.0 m×0.25 mm×0.25 μm)。升温程序:色谱柱初温80 ℃,保持3 min,然后以15 ℃/min升温至260 ℃,保持10 min。进样口温度:230 ℃。进样方式:不分流进样。进样体积:1 μL。载气:高纯氮气(纯度≥99.999%)。流速:1.0 mL·min-1。模式:恒压。
1.5 标准溶液的配制
称取适量的丙泊酚标准品于棕色容量瓶中,用乙酸乙酯溶解并定容至刻度,配成质量浓度为1.0 mg/mL的标准储备液,在-18 ℃条件下避光保存,有效期6个月。使用时用乙酸乙酯将储备液稀释成试验所需浓度的标准工作溶液,临用现配。
2 结果与讨论
2.1 溶剂的选择
试验采用气相色谱法测定丙泊酚麻醉剂残留量。使用的溶剂主要有乙酸乙酯-丙酮(1∶1,V/V)[10]、乙酸乙酯[11]和丙酮[7]三种。将丙泊酚标准储备液,分别用乙酸乙酯-丙酮(1∶1,V/V)、乙酸乙酯和丙酮稀释成10 μg/mL,进行气相色谱检测,色谱图如图1所示。结果显示,当乙酸乙酯-丙酮(1∶1,V/V)作为溶剂时,基线不平稳,有较大干扰峰出现;当乙酸乙酯作为溶剂时,无杂峰干扰,且基线平稳;当丙酮作为溶剂时,目标峰与内标峰附近基线不平稳,影响准确定量。所以,选择乙酸乙酯为最终溶剂。
2.2 色谱条件优化
2.2.1 柱温的选择
试验采用HP-50+毛细管柱,该类柱子的使用温度范围是40~280 ℃,因此柱温选择在正常使用范围内,按照温度梯度变化,选择70,80和90 ℃依次分析,色谱图见图2。结果发现,柱温的变化除了对保留时间有影响外,其对峰型不会有较大的影响,当柱温降低时,出现基线不平稳现象;当柱温升高时,峰形有拖尾现象。因此,为了考虑峰形,选择80 ℃作为试验柱温。

图1 三种不同溶剂比较色谱图
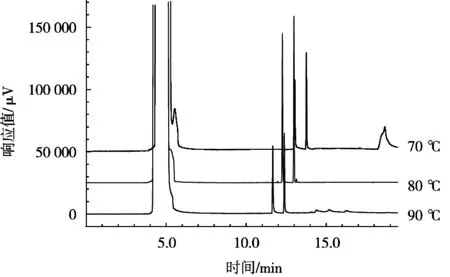
图2 不同柱温比较色谱图
2.2.2 进样口温度的设置
试验对进样口温度的考察结果如图3所示。结果表示进样口温度对峰形没有太大的影响。从不同进样口温度下相同浓度的标准物质的保留时间对比可知,不同进样口温度对保留时间也没有太大影响,考虑到仪器使用寿命,选择较低温度作为试验温度,因此选择230 ℃为进样口温度。
2.3 净化条件的选择
由于虾肉、鱼肉等水产品中含有大量的蛋白质、脂肪等物质,其基质特别复杂[12],对于丙泊酚的净化方法报道较少,所以试验采用不同固相萃取方式进行样品净化。
2.3.1 ProElut PLS固相萃取柱净化
黄武等[13]采用ProElut PLS固相萃取柱净化,高效液相色谱法测定丁香酚类化合物。因此,在此试验基础上,尝试使用ProElut PLS的方法进行净化,其操作步骤是:样品经丙酮提取后,将上清液移至10 mL的试管中,用3 mL甲醇、水活化ProElut PLS固相萃取柱,加入3 mL试样,然后用5%的甲醇水淋洗,抽干,3 mL甲醇洗脱,洗脱液用氮吹仪吹至尽干,用乙酸乙酯复溶至1 mL,过滤膜,待气相测定。结果表明,通过PLS净化过的样品在目标峰出现的时候,无明显杂质影响,因此对其进行了加标试验,其色谱图见图4。结果显示,虽然在空白样品净化时,杂峰对样品的出峰没有明显干扰,但是在加标回收试验中,发现其在目标物和内标物出峰时间未出峰,时间略有延迟,检测到的峰不是目标峰所在保留时间,因此此方法不适用。
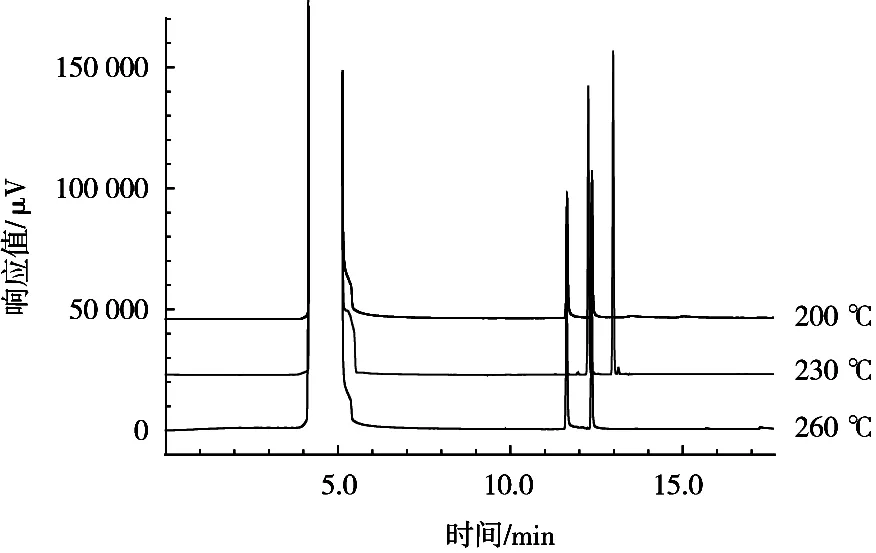
图3 不同进样口温度比较色谱图
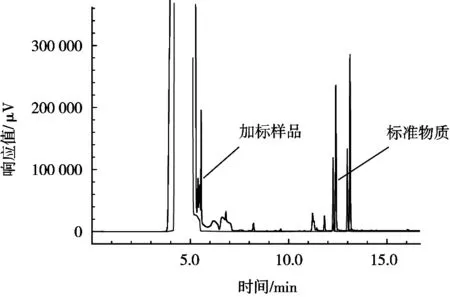
图4 ProElut PLS柱加标色谱图
2.3.2 C18反相固相萃取柱净化
C18反相固相萃取柱是以强疏水性硅胶基体为吸附剂,对非极性化合物具有较好的保留性,目前较为广泛应用[14]。试验尝试使用C18反相固相萃取柱的方法进行净化,其操作步骤是:样品经丙酮提取后,将上清液移至10 mL的试管中,加入200 μL四甲基氢氧化铵,吹至尽干,然后用乙酸乙酯复溶至5 mL。将C18柱置于固相萃取装置,用5 mL甲醇、水活化,加入5 mL试样,然后用5%的甲醇水淋洗,抽干,5 mL甲醇洗脱,洗脱液用氮吹仪吹至尽干,用乙酸乙酯复溶至1 mL,过滤膜,待气相测定。结果表明,在目标峰出现的时候,无明显杂质影响,因此对其进行加标试验,其色谱图见图5。结果显示,虽然在空白样品中,杂质峰的影响很小,但是在进行加标试验后,并未出现目标峰,回收率低,无法满足检测条件,因此C18反相固相萃取柱方法不可取。

图5 C18净化加标色谱图
2.3.3 EMR-Lipid结合EMR-Polish反萃取管净化
EMR-Lipid增强型脂质去除产品和EMR-Lipid Polish反萃管具有出色的基质净化效果,能够去除大部分基质(尤其是脂质),且不会对分析物回收率造成显著影响[15]。其操作步骤是:样品经10 mL丙酮提取后,上清液待净化。取15 mL EMR-Liplid dSPE分散净化管,加入5 mL水,摇匀,然后吸取5 mL待净化液至净化管中,旋涡振荡1 min,充分混匀,离心机以5 000 r/min离心3 min,将离心后的提取液移取至EMR Lipid Polish反萃取管中,旋涡混合,吸取1 mL净化液于进样小瓶中,待气相分析。结果表明,EMR-Lipid结合EMR-Polish反萃取管净化的空白样品在目标峰出现时未有杂峰影响,因此对其进行了加标试验,其色谱图见6。结果显示,经过净化的样品目标峰、内标峰完全与杂峰分离,对其进行的加标回收试验回收率较高,经过多次加标发现其重现性较好。因此,试验选择了这种净化方式。
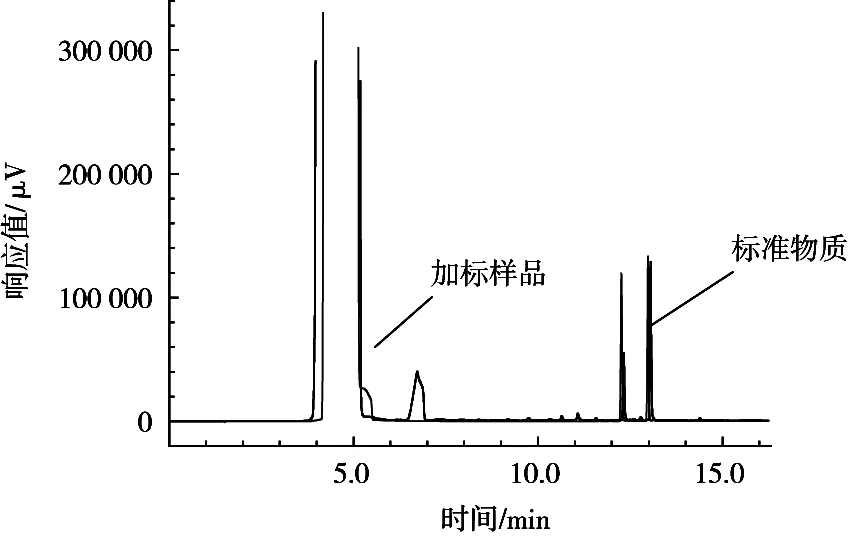
图6 EMR-Lipid结合EMR-Polish反萃取管加标净化加标色谱图
2.4 加标回收率
在南美白对虾、鳜、金鲳中分别添加质量浓度为1.0,2.0和10.0 μg/mL水平的标准溶液,内标物质量浓度为40.0 μg/mL,同时设置一个空白,计算丙泊酚麻醉剂的加标回收率及相对标准偏差(RSD),结果见表1。结果表明,当丙泊酚添加质量浓度为1.0~10.0 μg/mL时,其加标回收率为83.75%~102.18%,相对标准偏差为1.54%~7.39%,表明该方法的准确性和重现性好,符合残留分析要求。

表1 丙泊酚加标回收率
2.5 方法的线性范围
根据1.4色谱条件,将1.0 mg/mL的丙泊酚标准储备液用乙酸乙酯稀释成0.4,0.8,1.0,2.0,4.0,8.0和10.0 μg/mL的丙泊酚标准工作液,然后将1.0 mg/mL的百里香酚标准储备液用乙酸乙酯稀释成4.0 μg/mL的百里香酚内标标准工作液进行分析,以丙泊酚和百里香酚的浓度比为横坐标,峰面积比为纵坐标,做回归方程。结果表明,以该方法分析,丙泊酚在10.0~100.0 μg/mL质量浓度范围内其线性关系良好,线性回归方程为Y=1.198 2X-0.050 4,R2=0.999,检出限为0.3 mg/kg。
2.6 方法的重现性和稳定性
取同一浓度的丙泊酚标准溶液,共5份,分别连续进样后,根据峰面积测定,RSD值为1.3%,小于5%,表明该方法的重现性良好;同时将标准溶液置于0,3,9,12和24 h,测定其含量,RSD值为2.5%,表明该方法的稳定性良好。
3 结论
通过前处理方法的优化,样品用乙酸乙酯提取后,使用EMR-Lipid结合EMR-Polish反萃取管净化,经过0.25 μm的滤膜过滤后,用气相色谱仪检测。通过优化色谱条件,确定色谱条件为HP-50+毛细管柱,FID检测器,色谱柱温度80 ℃、进样口温度230 ℃、检测器温度280 ℃,升温程序:色谱柱初温80 ℃,保持3 min,然后以15 ℃/min,至260 ℃,保持10 min。进样模式为不分流,进样量1 μL,流速1 mL/min。水产品中丙泊酚质量浓度在0.4~10.0 μg/mL范围内线性关系良好,样品在加标质量浓度为0.4~10.0 μg/mL范围内,其平均加标回收率为83.75%~102.18%,相对标准偏差为1.54%~7.39%,检出限为0.3 mg/kg。试验所建立的检测方法适用于水产品中丙泊酚残留的测定。
