应用酵母双杂交技术筛选E种肠道病毒HY12-VP1蛋白的互作蛋白
2020-03-13郑博伟刘亚静张泽财王玮玉蔡梦露王浴光王新平吉林大学动物医学
郑博伟,刘亚静,张泽财,衣 帆,王玮玉,蔡梦露,王浴光,王新平,2∗ (.吉林大学 动物医学
学院,吉林 长春130062;2.教育部人兽共患病研究重点实验室 吉林 长春130062)
酵母双杂交系统是利用酵母转录因子的相关功能特性,通过2个重组蛋白在酵母细胞中因相互作用而靠近,进而引起下游报告基因的激活来捕获新蛋白的一种试验方法。1989 年,FIELD 和SONG等[14]首次提出酵母双杂交系统。真核生物转录因子GAL4包含2个结构上互相独立但功能上却息息相关的结构域:DNA 结合结构域(DNA-binding domain,BD)和转录激活结构域(transcription-activating domain,AD)。BD 可和上游激活序列(upstream activating sequence,UAS)特异性结合,AD可以激活UAS下游的基因。但是单独的BD 或AD都不能发挥作用,只有BD 和AD 因相互作用而发生空间上的靠近才能激活下游的报告基因,进而通过缺陷型培养基和显色反应即可筛选到阳性菌落。
BEV 感染是近年来国内新发传染病,对其致病机理及如何诱发机体免疫应答的研究鲜有报道[7]。本试验以E种肠道病毒HY12毒株编码的VP1蛋白为诱饵蛋白,利用酵母双杂交系统对MDBK 细胞的cDNA 文库进行筛选,初步发现与HY12-VP1相互作用的潜在蛋白,为后续研究VP1 蛋白在BEV感染过程中发挥的作用奠定基础。
1 材料与方法
1.1 病毒、细胞及载体HY12病毒株为本实验室从长春某牛场暴发的1起疫病中首次分离到的1株E种肠道病毒,-80℃保存。牛肾细胞(MDBK)、E.coliTOP10 细胞由本实验室保存。酵母菌株AH109、Y187,质粒p GBKT7、p GADT7、p GBKT7-P53、pGBKT7-Lam 由吉林大学动物医学学院寄生虫实验室惠赠。大肠杆菌E.coliTOP10感受态由本实验室制备,-80℃保存。
1.2 主要试剂DNA 胶回收试剂盒、质粒小量提取试剂盒、酵母质粒小量提取试剂盒、酵母转化试剂盒、YPDA、SD/-Leu、SD/-Trp、SD/-Leu/-Trp、SD/-His/-Leu/-Trp、SD/-Ade/-His/-Leu/-Trp、3AT 购自北京天根生化科技有限公司;限制性内切酶Bam HⅠ、EcoRⅠ、rTaq酶、T4DNA 连接酶等购自大连宝生物工程有限公司;PEG3350、DMSO、X-αgal、鲑鱼精DNA、Li Ac、YNB 及各种氨基酸购自Sigma公司;异丙醇、无水乙醇、氯仿均为国产分析纯产品;DMEM 购自美国Invitrogen公司;胎牛血清购自上海克隆生物化学制品有限公司;Reverse Transcriptase M-MLV 试剂盒购自Ta KaRa公司。
1.3 方法
1.3.1 细胞与病毒的培养 使用含有5%胎牛血清(FBS)的DMEM 培养液培养MDBK 细胞。当细胞生长至70%~80%时,接种HY12病毒液并培养至50%~70%细胞出现病变。
1.3.2 RNA 提取与PCR 扩增VP1基因序列 以TRIzol试剂盒提取接毒细胞的总RNA,然后利用反转录试剂盒将提取到的RNA 反转录成c DNA。引物的设计参照HY12结构蛋白VP1的基因序列进行。引物由上海生工生物工程公司合成,
其次,在“绿色原则”作为物权人需承担环保义务的一般性条款在“物权编”总则中确立的情况下,“物权编”分则的制度构建及规范解释在强调物权之经济价值的同时,必须对物权之生态环境功能进行重新考量和定位,并在具体制度构建中尽力给出二者冲突时的解决规则和取舍标准。一方面,这需要将《物权法》中已蕴含“绿色原则”理念的私法规范进行规则整合,实现“‘绿色原则’规则化”,避免在个案中缺少基本规范而“向基本原则逃避”;另一方面,在承继《物权法》已蕴含“绿色原则”既有规范前提下,将更多蕴含此种理念的制度规范纳入《民法典》“物权编”中,使“绿色原则”转变为可操作的具体物权规范。
上游引物:5′-CGCGGATCCGCGTCTCTCATCGGTGG-3′;
下游引物:5′-CCGGAATTCCATCTGGTAGTTGGTCTCTAGTCA-3′,扩增的PCR 产物以1%琼脂糖进行鉴定。
1.3.3 酵母诱饵重组质粒的构建与鉴定 将PCR扩增的产物以EcoRⅠ和Bam HⅠ限制性内切酶进行双酶切,并将其连接到诱饵载体pGBKT7 的EcoRⅠ/Bam HⅠ酶切位点上。连接产物转化到Top10感受态细胞中,冰浴30 min,放入42℃水浴锅中热激70 s,冰浴4 min后,加入1 m L LB液体培养基,于37℃摇床振荡培养(200 r/min)1 h后,取100μL 菌液,均匀涂布在卡那霉素抗性的LB 琼脂平板上,37℃培养过夜。次日挑取单个菌落,进行培养,抽提质粒,然后利用EcoRⅠ和Bam HⅠ限制性内切酶进行双酶切鉴定。将鉴定后的重组质粒命名为p GBKT7-VP1。
1.3.4 AH109酵母感受态细胞的制备 从YPDA琼脂平板上挑取新鲜的AH109单克隆菌落至2 m L YPDA 培养基中,于30℃以200 r/min 振荡培养17 h 后,吸取1 m L 菌液,置于50 m L YPDA 液体培养基中,于30℃摇床中200 r/min 摇至D600>1.5。取适量菌液加入到300 m L YPDA 培养基中,培养至D600=0.5;3 000 r/min 离心收菌,加 入40 m L dd H2O 或TE重新洗涤酵母沉淀细胞,重复洗2次。最后以1 m L 1×TE/LiAc溶液悬浮沉淀菌体,即为酵母感受态细胞。
1.3.5 诱饵重组质粒转化AH109酵母菌株 取1个1.5 m L的EP管,加入0.1μg pGBKT7-VP1 和10μg预处理过的鲑鱼精DNA,混合均匀,再加入100μL的酵母感受态细胞,吹吸混匀,继续加入600μL PEG/LiAc,剧烈振荡混匀,于30℃、200 r/min 摇床中培养30 min。取出摇床中的EP 管,加入50μL DMSO轻柔混匀,于42℃水浴锅中加热15 min,冰浴3 min。离心收菌后,用0.5 m L 的TE 重悬酵母细胞。吸取150μL 菌液,均匀涂布在固体SD/-Trp培养平皿上,封口倒置在30℃酵母培养箱中培养1~3 d,p GBKT7空质粒作为对照组。
1.3.6 诱饵重组质粒的自激活活性及毒性检测 取30μL无菌水悬浮pGBKT7-VP1单菌落,分别接种 于SD/-Trp/-Leu、SD/-Trp/-Leu/-His/-Ade 培养平皿上。封口倒置在30℃酵母培养箱中,培养1 d观察结果,pGBKT7空质粒作为对照组。挑取单个菌落接种于4 m L 的YPDA 液体培养基中,置于30℃、200 r/min摇床中振荡培养,比较试验组和对照组不同时间菌液的D600值。
1.3.7 cDNA 文库滴度和丰度检测 吸取1 m L文库菌液依次做10的倍比稀释(101~105),分别取不同稀释倍数的菌液100μL,均匀涂布在SD/-Leu固体培养平皿上,封口后于30℃培养3~5 d,确定菌落数量,计算文库滴度。文库滴度按公式以N×稀释倍数/涂板菌液体积进行计算。从SD/-Leu固体培养平皿上随机挑选44 个菌落,分别溶于20μL dd H2O 中,反复冻融后以12 000 r/min离心5 min,取其上清液作为模板,并以文库通用引物进行PCR。
1.3.8 文库菌与诱饵菌的杂交 挑取单个新鲜培养的诱饵菌接种到50 m L SD/-Trp液体培养基中。于30℃、200 r/min摇床中振荡培养16 h至D600=0.8。离心收菌,弃掉上清,用4~5 m L SD/-Trp液体培养基重悬酵母细胞沉淀。将1 m L 文库菌和4~5 m L诱饵菌在无菌锥形瓶中混合。在锥形瓶中加入45 m L 2×YPDA 液体培养基混合均匀。用1 m L 2×YPDA 液体培养基冲洗文库管2次,冲洗液加入到2 L锥形瓶中。于30℃、30~50 r/min摇床中缓慢振荡培养20~24 h,直至出现三叶草结构状的结合子。在显微镜下观察到结合子出现时终止培养,离心收菌。用0.5×YPDA 液体培养基冲洗锥形瓶,重悬离心后收集到的细胞,弃上清。重复冲洗、离心的步骤2次,最后用10 m L 0.5×YPDA 液体培养基重悬细胞,将10 m L细胞悬液全部涂布于150 mm DDO 平板,每个平板均匀涂布200μL,封口倒置在30℃酵母培养箱中培养3~7 d。将DDO平板上长出的菌落点涂到QDO 平板上,封口倒置在30℃酵母培养箱中培养1~3 d。再将QDO 平板上长出的菌落点涂至QDO/X/A 平板上,封口倒置在30℃酵母培养箱中培养1~3 d。
1.3.9 酵母双杂交阳性克隆的鉴定 用酵母质粒小量提取试剂盒抽提所有在QDO/X/A 平板上变蓝的克隆。将抽提的酵母混合质粒转化至E.coliTOP10感受态细胞中,均匀涂布在氨苄抗性的LB琼脂板上,挑取单克隆,用质粒小量提取试剂盒抽提质粒,以质粒为模板进行PCR,阳性样品送公司测序,并将测序结果进行BLAST 比对分析,确定潜在的互作宿主蛋白。将抽提的阳性大肠杆菌质粒与构建的诱饵质粒p GBKT7-VP1 共同转化到酵母菌AH109中。设置阴性对照组、阳性对照组、试验组(p GBKT7-VP1和阳性质粒共转)。将转化后的菌液均匀涂布在DDO 平板上,封口倒置在30℃酵母培养箱中培养1~3 d。将DDO 平板上长出的菌落点涂到QDO 平板上,封口倒置在30℃酵母培养箱中培养1~3 d。再将QDO 平板上长出的菌落点涂至QDO/X/A 平板上,封口倒置在30℃酵母培养箱中培养1~3 d,观察结果。
2 结果
2.1 pGBKT7-VP1的构建与酶切鉴定利用1%琼脂糖凝胶电泳来观察基因扩增结果,发现PCR 扩增出与预期大小相符的846 bp目的片段(图1A)。挑取5个转化后的菌落,利用质粒小量提取试剂盒抽提质粒,然后用限制性内切酶Bam HⅠ和EcoRⅠ对构建的重组质粒进行双酶切,经1%琼脂糖凝胶电泳检测,出现1 条846 bp 左右的目的条带和1 条7 300 bp左右的pGBKT7载体片段即为阳性质粒(图1B),结果显示2 号质粒为构建成功的p GBKT7-VP1诱饵质粒,将质粒送至上海生工生物工程公司测序,进一步确定质粒构建成功。

图1 pGBKT7-VP1的构建与酶切鉴定 A.VP1基因的扩增结果;B.重组质粒的双酶切鉴定;Ma.DL2000 DNA Ladder Marker;N.VP1 基因;Mb.DL15000 DNA Ladder Marker;1~5.构建的重组质粒酶切样品
2.2 重组质粒pGBKT7-VP1对酵母细胞的毒性验证对分别含有p GBKT7-VP1和p GBKT7质粒的AH109菌株的D600值进行测定,结果菌株的D600值在不同培养时间点无明显差异(图2),表明重组质粒p GBKT7-VP1对酵母细胞AH109无毒性作用。
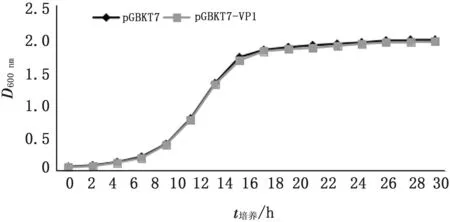
图2 包含不同质粒的AH109酵母细胞生长曲线
2.3 重组质粒pGBKT7-VP1的自激活活性验证将p GBKT7-VP1 重组质粒转化到酵母AH109 菌株中,均匀涂布在SD/-Trp固体平皿上,挑取单菌落,用30μL 无菌水充分悬浮,涂布在固体SD/-Trp/-Leu、SD/-Trp/-Leu/-His/-Ade培养平皿上。封口倒置在30℃酵母培养箱中培养1~2 d。结果如图3所示,菌株在SD/-Trp培养平皿上可以正常生长,而在SD/-Trp/-Leu、SD/-Trp/-Leu/-His/-Ade培养平皿上均没有生长,说明重组质粒没有自激活活性。
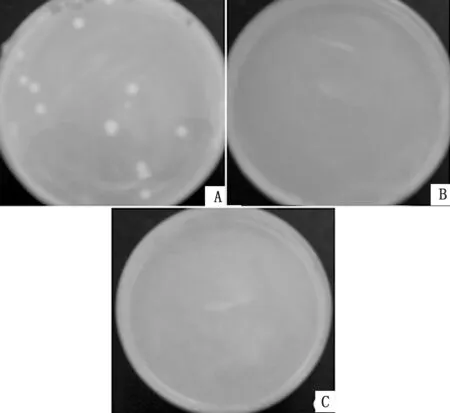
图3 诱饵质粒的自激活活性检测 A.SD/-Trp固体培养皿;B.SD/-Trp/-Leu 固 体 培 养 皿;C.SD/-Trp/-Leu/-His/-Ade固体培养皿
2.4 cDNA 文库滴度与丰度检测将文库菌液做10的倍比稀释后,取各个稀释度菌液100μL,涂布在SD/-Leu固体平皿上,计算每个滴度生长的酵母克隆数。通过计算,确定文库滴度为108CFU/m L。随机挑取平皿上44个克隆进行PCR扩增检测,结果如图4所示,插入片段大小范围为200~2 000 bp,说明文库多态性良好,丰度较高,符合试验要求,可以进行后续试验。
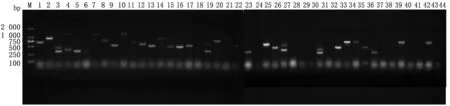
图4 cDNA 文库插入片段电泳图 M.DL2000 DNA Ladder Marker;1~44.随机挑取的单克隆
2.5 阳性克隆的筛选将含有MDBK 细胞基因组文库的菌株与含有诱饵质粒的菌株混合培养后,进行酵母转化试验,培养16 h后,取菌液滴于载玻片。显微镜观察到大量米奇头样(图5)的结合子,表明杂交成功。
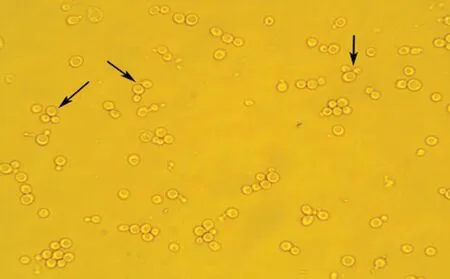
图5 酵母双杂交中的结合子
文库菌与诱饵菌杂交后,于QDO 培养皿中培养72 h后,将生长的菌落点涂至QDO/X/A 培养皿上,直至克隆变蓝(图6)。
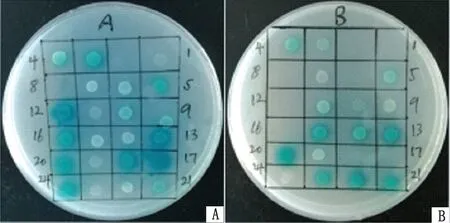
图6 QDO/X/A 培养皿检测筛选结果
2.6 阳性酵母克隆质粒的PCR 及测序鉴定将显色变蓝的克隆进行质粒的抽提,并以质粒为模板进行PCR,结果扩增出大小不一的片段(图7)。对扩增片段进行测序测定与分析,获得12 个候选蛋白(表1)。

图7 阳性酵母克隆质粒的PCR 结果 M.DL2000 DNA Ladder Marker;1~12.阳性酵母克隆
3 讨论
BEV 属于小RNA 病毒科,其编码的前体多聚蛋白经降解可产生VP1、VP2、VP3和VP4 4种结构蛋白。结构蛋白VP1~VP3是病毒最先编码的3个蛋白,含有中和抗体的特异性结合位点。研究表明,VP1~VP3与病毒感染和病毒血清型的分类密切相关[15-17]。结构蛋白VP1位于病毒粒子的表面,带有大量的中和抗原决定簇,直接决定着病毒的抗原性,在感染宿主细胞过程中,发挥了极其重要的作用[18-19]。目前,有关VP1蛋白功能的研究主要集中于小RNA 病毒科中脊髓灰质炎病毒、EV71、口蹄疫病毒等,但有关BEV 致病机理的研究较为缺乏。
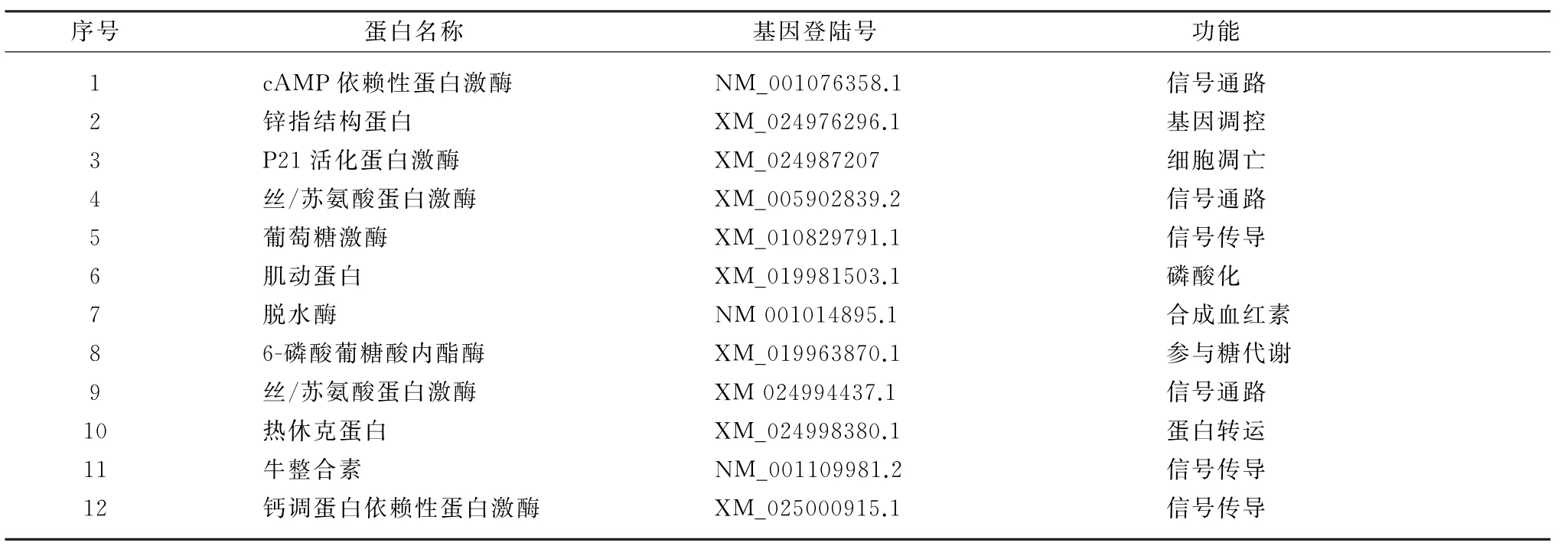
表1 筛选出HY12-VP1的潜在互作蛋白基因
目前,筛选互作蛋白的方法众多,但由于酵母双杂交技术具有简单、高效、易于操作等优点,因此本研究选择该方法来筛选HY12毒株VP1蛋白的互作蛋白。将真核生物转录因子的2个结构域与不同蛋白进行融合表达:BD 与诱饵蛋白融合表达,即构建诱饵重组蛋白的质粒;AD 与文库蛋白融合表达,即构建文库重组蛋白的质粒[20],将这2种重组质粒同时转化酵母菌株AH109 中。由于酵母菌株AH109在正常情况下不能合成亮氨酸(Leu)、色氨酸(Trp)、组氨酸(His)与腺嘌呤(Ade),因此无法在缺乏这些营养物质的培养基上生长,只有当诱饵重组蛋白与文库重组蛋白因相互作用而在空间上靠近时,BD 和AD 在空间上也相互靠近从而使得转录因子具有转录活性,进而激活下游报告基因HIS3、LACZ等的表达[21]。
本试验应用酵母双杂交技术,初步筛选了HY12-VP1蛋白的互作的蛋白。成功构建了重组诱饵质粒pGBKT7-VP1并经试验验证该重组诱饵质粒无自激活活性和细胞毒性。将重组质粒的酵母菌株与文库菌株进行混合培养,初步筛选到12个阳性克隆,即候选蛋白。这些候选蛋白的功能各不相同,如钙调蛋白依赖性蛋白激酶(Ca MK)是一种依赖于钙/钙调蛋白(calmodulin,Ca M)的丝氨酸/苏氨酸蛋白激酶,在脑组织中参与神经信号的传导[22];热休克蛋白(heat shock proteins,HSPs)在蛋白质折叠、装配、加工等多种生物水平的活动中发挥了极其重要的作用[23]。热休克蛋白表达量的上升,则可以通过影响癌细胞在基因水平上的转录,从而提高癌细胞的黏附能力浸润现象的发生[24]。在候选蛋白中,P21 活化蛋白激酶1(p21-activated protein kinases,PAK1)属于丝氨酸苏氨酸蛋白激酶家族[25]。据报道,多种刺激因子可以激活PAK1,从而引起下游信号分子的活化,导致细胞凋亡[26],进而影响病毒的毒力和致病性。本研究初步确定出VP1与PAK1互作,表明PAK1很可能通过调控细胞凋亡影响病毒的复制,但具体机制尚不清楚,有待进一步研究。此外,ZC3H11A 即CCCH 型锌指结构蛋白(Zinc-finger Antiviral Protein,ZAP)可以特异性的与靶点结合来识别核酸和蛋白质、调控基因的转录和蛋白质与蛋白质间的相互作用[27]。ZAP在宿主细胞内可发挥抗病毒作用。首先,ZAP可与靶病毒m RNA 的特异性序列结合,使得宿主细胞的核酸外切酶复合体Exosome 从3′端降解病毒的m RNA[28],同时,ZAP 还可以利用p72(RNA 解旋酶)去除病毒m RNA 上的帽子结构[29]。其次,在病毒翻译的过程中,eIF4A 翻译起始因子和eIF4G 翻译起始因子会产生互作,ZAP却与eIF4A 结合来影响eIF4A 和eIF4G 的互作,进而抑制了病毒m RNA的翻译,一定程度上起到了抗病毒感染的作用[30-31]。因此,我们预测过表达ZAP可以在一定意义上减少病毒感染。综上所述,本试验应用酵母双杂交技术筛选出12个VP1的可能互作蛋白,为后续肠道病毒感染机制及诱发机体免疫的相关研究提供了基础。
