猪伪狂犬病病毒野毒和猪圆环病毒3型双重PCR检测方法的建立
2020-03-13崔建涛田润博韩昊莹郑慧华陈红英河南农业大学牧医工程学院河南
赵 宇,崔建涛,田润博,韩昊莹,郑慧华,刘 芳,陈红英,2∗ (.河南农业大学 牧医工程学院,河南
郑州450002;2.郑州市猪重大疫病防控重点实验室,河南 郑州450002)
伪狂犬病(pseudorabies,PR)是由伪狂犬病病毒(pseudorabies virus,PRV)引起的多种家畜和野生动物以发热、奇痒(猪除外)及脑脊髓炎为主要症状的一种重要传染病[1]。PRV 属于疱疹病毒科,α-疱疹病毒亚科,其基因组大小约为150 kb,可以编码多种病毒蛋白的双链DNA 病毒[2]。PRV 能够感染各个年龄阶段的猪只,仔猪感染后死亡率达到100%,育肥猪感染后生长缓慢,妊娠母猪感染后发生繁殖障碍,出现流产,产死胎,木乃伊胎等现象,给我国养猪业带来了巨大的经济损失[3]。
2016年,PALINSKI等[4]从患病母猪和流产胎儿体内首次鉴定出猪圆环病毒3型(porcine circovirus type 3,PCV3)。PCV3为单股环状的DNA 病毒,属于圆环病毒科,圆环病毒属,PCV3主要包括2个开放阅读框(ORF),Rep基因与病毒的复制相关,编码297个氨基酸,Cap蛋白主要与病毒的致病性相关。PCV3与皮炎与肾病综合征、呼吸道疾病以及繁殖障碍等疾病有关,在死胎和精液中可以检测出PCV3[5-6],已引起国内外兽医工作者的广泛关注。目前PCV3在美国[4]、中国[5]、巴西[6]、泰国[7]、韩国[8]、日本[9]以及欧洲等多个国家和地区流行[10-15]。
PRV 与PCV3的混合感染在我国猪场已经存在[16],且PRV 和PCV3 均可引起母猪繁殖障碍[3-5],在临床诊断上难以区分这2种病原体引起的感染,因此需要建立实验室的诊断方法。目前,PRV 的检测方法有病毒分离、ELISA、普通PCR 和qPCR 等检测技术,PCV3的检测方法有普通PCR、qPCR 和ELISA 等,然而迄今尚未有建立能够同时检测和区别PRV 和PCV3的检测方法。因此,本试验建立的能够同时检测PRV 和PCV3的双重PCR检测方法,为我国PRV 和PCV3的防控和检测提供技术支持。
1 材料与方法
1.1 病毒株、菌株、临床样品通过PCR 方法对郑州市猪重大疫病防控重点实验室保存的病料进行PCV3扩增并测序,将鉴定为阳性的病料作为PCV3阳性对照。PRV 强毒株(PRV HN2012)由郑州市猪重大疫病防控重点实验室从患有神经症状的仔猪中分离并保存。PCV2、猪细小病毒(PPV)、猪蓝耳病病毒(PRRSV)、猪瘟病毒(CSFV)、猪流行性腹泻病毒(PEDV)均由郑州市猪重大疫病防控重点实验室保存。
2017-2018 年,39 份病料样品(包括脑,淋巴结,小肠,肺脏和脾脏)采自山西省文水,河南省孟州、新密以及许昌等10个地市14个养殖场患有繁殖障碍疾病的39头猪。取适量的样品,制成组织匀浆,经-80℃反复冻融3 次后,12 000 r/min 离心5 min。取上清液,于-80℃保存备用。
1.2 主要试剂TRIzol和反转录试剂盒均购自南京诺唯赞生物科技有限公司;病毒DNA 快速纯化试剂盒和凝胶回收试剂盒均购自生工生物工程(上海)股份有限公司;2×TaqMaster Mix购自北京康为世纪生物科技有限公司;p MD18®-T 载体、X-gal、IPTG、琼脂糖等均购自宝生物工程(大连)有限公司;其他常规试剂均为分析纯。
1.3 引物设计参考GenBank 中登录的PRV(KF042383、KF017615、KF130880)g E基 因 和PCV3的全基因组序列(KX778720、KX966193),利用引物软件Primer 5.0,设计了2对特异性引物,预期扩增片段长度分别为PRV 的429 bp和PCV3的344 bp。引物由上海生工生物工程技术服务有限公司进行合成(表1)。

表1 双重PCR 引物序列
1.4 核酸的提取按照病毒DNA 快速纯化试剂盒说明书,分别进行PRV、PCV3、PCV2、PPV DNA的提取,并于-80℃保存;根据TRIzol说明书提取PEDV、PRRSV 和CSFV 总RNA,按照反转录试剂盒说明书合成cDNA,并于-80℃保存备用。
1.5 PRV和PCV3阳性标准样品的制备利用引物P1和P2 分 别 进 行PRV 和PCV3 单 项PCR 扩增。反应体系为:2×TaqMaster Mix 10 μL,dd H2O 8μL,上、下游引物各为0.5μL,模板量为1.0μL。反应程序为95℃5 min;95℃30 s、56℃25 s,72℃30 s,共35 个循环;72℃10 min。PCR产物经3%琼脂糖凝胶电泳进行检测。用DNA 凝胶回收试剂盒回收PCR 产物,纯化后连接至p MD18-T 载体、构建重组质粒p MD18-PRV 和p MD18-PCV3。将鉴定为阳性的重组质粒进行序列测定,且与GenBank中选取的参考序列进行同源性比对,以验证扩增产物的特异性。利用紫外分光光度计检测2种阳性质粒浓度并分别计算拷贝数。
1.6 PRV和PCV3双重PCR的建立
1.6.1 双重PCR 方法最佳条件 通过对退火温度(52~62℃)、引物比例进行优化,最终确定双重PCR 的最佳反应条件。
1.6.2 双重PCR 的特异性试验 以PRV、PCV3、PCV2、PRRSV、PEDV、PPV、CSFV 的DNA 或cDNA 为模板,同时设立阴性对照,根据已优化好的双重PCR 条件进行扩增,以检测该方法的特异性。
1.6.3 双重PCR 的灵敏度试验 将已构建的重组质粒p MD18-PRV 和p MD18-PCV3等体积混合后进行10倍倍比稀释,并将各个稀释度的标准质粒作为模板通过已优化条件的双重PCR 方法进行扩增,检测该方法的灵敏度。
1.7 双重PCR的重复性试验通过已优化条件的双重PCR 方法对采自不同地区含PRV 和PCV3的3份阳性病料样品DNA 进行双重PCR 检测,并做3次重复。
1.8 临床样品的检测利用双重PCR 方法对采自山西省文水,河南省孟州、新密以及许昌等10个地市14 个养殖场的39 份病料进行检测,并与单项PCR 检测方法进行比较。
2 结果
2.1 单项PCR 扩增结果用临床组织样品DNA作为模板,利用引物P1和P2 分别进行PCR 扩增PRV 和PCV3,扩增大小分别为429和344 bp(图1),与预期相符。将PCR 产物纯化回收后,连接到载体p MD18-T 中进行测序,测序结果表明,所克隆的PRV 目的片段大小为429 bp,与GenBank中的PRV 参考株核苷酸序列同源性为100%;所克隆的PCV3目的片段大小为344 bp,与GenBank 中的PCV3参考株核苷酸序列同源性为100%,表明PCR 产物为PRV 和PCV3的目的片段。通过紫外分光光度计分别检测PRV 和PCV3的浓度,经计算PRV 的拷贝数为5.02×1010copies/μL,PCV3的拷贝数为9.12×1010copies/μL。

图1 PRV 和PCV3目的基因PCR 扩增结果 M.DL2000 DNA Marker;1.PRV 的PCR 产 物;2.PCV3 的PCR产物;3.PRV 的阴性对照;4.PCV3的阴性对照
2.2 双重PCR扩增结果通过反应体系和扩增条件的优化,最终确定双重PCR 的反应体系为2×TaqMaster Mix 10 μL,dd H2O 6 μL,PRV 和PCV3上、下游引物均为0.5μL,PRV 和PCV3 的混合DNA 为2.0μL。双重PCR 的反应程序为95℃5 min;95℃30 s、56℃25 s,72℃30 s,共35个循环;72℃10 min(图2)。
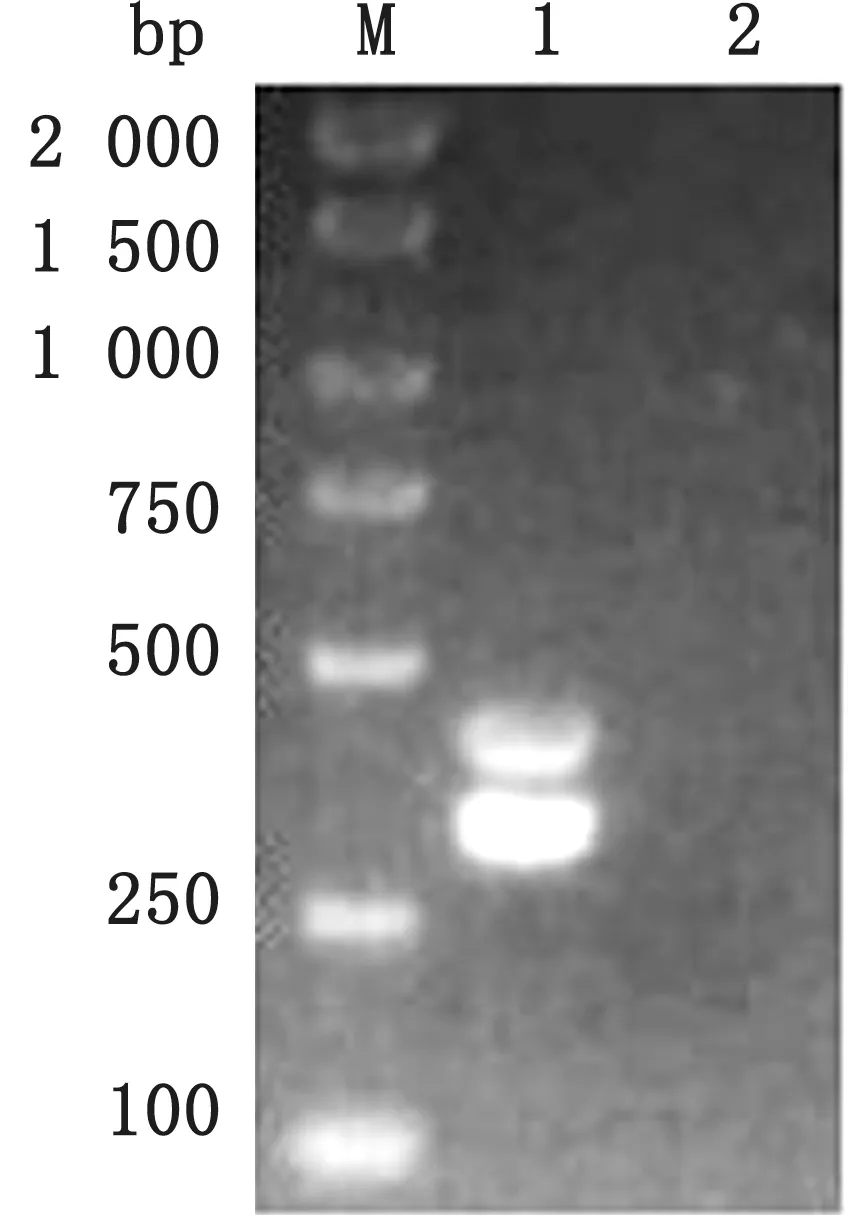
图2 PRV 和PCV3双重PCR 扩增结果 M.DL2000 DNA Marker;1.PRV 和PCV3 双 重PCR 产 物;2.PRV 和PCV3双重扩增阴性对照
2.3 双重PCR特异性结果通过已经优化条件的双重PCR 对PRV、PCV3、PCV2、PRRSV、PEDV、PPV、CSFV 的DNA 或c DNA 进行PCR 扩增,并设立阴性对照。同时可以扩增出PRV 的429 bp片段和PCV3的344 bp片段,而其他病毒核酸以及阴性对照均无特异性条带,表明该方法具有良好的特异性(图3)。

图3 双重PCR 特异性检测结果 M.DL2000 DNA Marker;1.双 重PCR 产 物;2.PCV2的PCR 产 物;3.PPV 的PCR 产 物;4.PEDV 的PCR 产 物;5.PRRSV DNA 模板的PCR 产物;6.CSFV DNA 模板的PCR 产物;7.阴性对照
2.4 双重PCR 灵敏度结果将PRV 和PCV3的标准阳性质粒等体积混合后进行10倍系列稀释,选取101~1010的稀释度进行双重PCR 扩增,同时设立阴性对照。结果显示,PRV 和PCV3的最低检测值分别为:502.0 和91.2 copies/μL,表明该方法具有良好的灵敏性(图4)。

图4 双重PCR 灵敏度结果 M.DL2000 DNA Marker;1~10.PRV 和PCV3混合模板作101~1010稀释后的扩增结果;11.阴性对照
2.5 双重PCR重复性试验结果通过已优化条件的双重PCR 方法对采自不同地区含PRV 和PCV3的3份阳性病料样品DNA 进行双重PCR 检测,并做3 次重复,每次PCR 扩增结果均为PRV 和PCV3阳性,表明该方法具有很好的可重复性。
2.6 临床诊断结果通过建立的双重PCR 和单重PCR 对采集的39 份临床样品进行PCR 扩增。扩增结果显示,PRV 的阳性率为17.95% (7/39),PCV3的阳性率为33.33% (13/39),混合感染率为10.26% (4/39),且双重PCR 与单重PCR 的结果相一致。
3 讨论
g E基因作为伪狂犬病病毒毒力基因之一,在PRV 的神经嗜性和毒力方面起到了重要的作用,但其编码的糖蛋白非病毒复制所必需,病毒缺失g E基因后依然能复制,因此检测g E基因来区分PRV野毒株感染和疫苗株感染[17]。自PCV3被发现以来,PCV3在世界范围内逐渐流行[4-15],并在我国多个省份多个猪场存在,给我国养猪业带来了潜在的威胁,有报道称PCV3有可能与繁殖障碍相关[4-5]。由于PRV 与PCV3均可引起繁殖障碍,在临床诊断上区分这2种病原体具有一定的难度,而双重PCR技术不仅在单重PCR 技术基础上能够满足对疾病混合感染的诊断,节约试验成本,而且更符合对疾病快速诊断的要求。因此,本试验针对PRV 的g E基因和PCV3 的Rep基因,建立能够同时检测PRV和PCV3 的双重PCR 检测方法,为检测和防控PRV 和PCV3提供技术支持。
本试验通过对双重PCR 的条件进行优化,最终建立最佳的双重PCR 检测方法。扩增结果显示,可以扩增出大小分别为PRV 429 bp 和PCV3 344 bp的特异性片段,2种扩增产物大小相差约80 bp,因此,利用凝胶电泳能够较好的、直观的分辨这2种病原体。特异性试验表明,除了PRV 和PCV3出现目的条带以外,其他病原体均无条带,表明该方法具有较高的特异性。灵敏度显示,PRV 的最小检测下线为502.0 copies/μL,与赵丽等[18]建立的PRV qPCR方法的灵敏度相当;PCV3 的最小检测下线为91.2 copies/μL,与肖琦等[19]、徐朋丽等[20]所建立的普通PCR 方法以及郝占武等[21]建立的PCV3 qPCR 方法检测下线相当,说明本试验所建立的双重PCR 检测方法具有良好的灵敏度。
本试验通过已经建立的双重PCR 检测方法,对采自山西省文水,河南省孟州、新密以及许昌等10个地市14个养殖场的39份病料进行PCR 扩增,结果显示PRV 的阳性样品数为7 份,阳性率为17.95% (7/39),其中4份阳性样品分布于河南省,3份阳性样品分布于山西省,表明在河南以及山西地区的猪场仍然存在着PRV 野毒的感染;PCV3的阳性样品数为13份,阳性率为33.33%(13/39),其中7份阳性病料分布于河南省,6份阳性病料分布于山西省,表明PCV3感染已经在河南省和山西省存在,并且阳性率与徐朋丽等[20]检测结果基本一致。PRV 和PCV3的混合感染样品数为4份,阳性率为10.26% (4/39),其中2份阳性病料分布于河南省,另外2份分布于山西省,表明PRV 和PCV3的混合感染已经出现在河南省以及山西省的猪场。
目前,虽然PCV3并未分离出来,但有研究报道PCV3与繁殖障碍有关,并且相关文献报道PCV3与PRV 的混合感染是存在的,由于PRV 与PCV3均有可能引起繁殖障碍,在临床诊断上难于区分这2种病原体。因此本试验建立一种快速,高效,简便的PRV 和PCV3 双重PCR 检测方法,为我国的PRV 和PCV3的防控和检测提供技术支持。
