水液相环境下氢氧根催化天冬氨酸手性对映体转变及质子的作用*
2020-02-26乔朝阳闫红彦孙永清刘一司吕路瑶佟华王佐成
乔朝阳,闫红彦,孙永清,刘一司,吕路瑶, 佟华,王佐成
(1.白城师范学院计算机科学学院,吉林 白城 137000;2.白城技师学院基础部,吉林 白城 137000;3.白城师范学院物理学院,吉林 白城 137000)
天冬氨酸(aspartic,Asp)是构成蛋白质的重要氨基酸,根据其对平面偏振光不同的旋光作用,分为左旋天冬氨酸(L-Asp)和右旋天冬氨酸(D-Asp);根据构型不同,分为S-构型(S-Asp)和R-构型(R-Asp)。L-Asp在生命体内具有活性,具有使蛋白质稳定的作用,临床上L-Asp盐酸盐用于治疗肝功低下和放射性药物中毒等,在食品工业用于保鲜剂、油脂抗氧剂和增香剂等[1]。D-Asp对大肠杆菌具有较强的抑制作用[2],还用于酒精中毒的治疗[3],用作手性药的中间体及生产具有消炎、抗皱、祛斑等功能的化妆品[4],还用于头孢类抗生素的合成原料等[5]。文献[6]的实验研究表明,有微量的D-Asp存在于生命体,但其来源尚不清楚。
基于光学纯的Asp有着不同的功效及D-Asp在生命体内的存在,为了在实验上实现廉价的L-Asp向昂贵的D-Asp异构,以及解释生命体内D-Asp的来源,人们对其旋光异构进行了研究。文献[7]的研究表明,通常的Asp具有稳定性,气相Asp的手性转变可在4个通道实现,其中以氨基氮为氢转移桥梁的通道具有优势。文献[8]采用长程校正泛函CAM-B3LYP研究了Asp的一种构象在水液相环境下的旋光异构,粗浅地解释了Asp的一种构象在生命体内的消旋。文献[9]采用色散校正密度泛函的ωB97X-D方法,研究了Asp的两种稳定构象的手性对映体转变及水分子簇的催化作用,结合自洽反应场的SMD模型方法,研究了水液相环境下水分子簇催化作用,解释了生命体内D-Asp几种稳定构象的来源。
生命体内有氢氧根离子(OH-)存在,数量过多会影响健康[10],但机理并不清楚。已有研究[11-12]表明,水液相环境下氢氧根(水分子簇)可以催化α-丙氨酸和缬氨酸旋光异构。已有研究[13]表明,碱处理的蛋白质中的Asp可以消旋,但反应机理尚不清楚。基于此,本工作对标题反应进行了研究。
1 研究与计算方法
考虑到Asp分子内以及氢氧根、水分子和Asp分子之间的氢键等长程作用,采用色散校正密度泛函ωB97X-D[14]方法,选用6-311++G(d, p)基组,结合自洽反应场理论的SMD模型[15]方法,在隐式溶剂模型下全优化标题反应的稳定点和过渡态[16],同时获得各个驻点零点振动能。通过IRC(内禀反应坐标)计算[17],确认过渡态的可靠性。为获得高水平的反应过程势能面,采用微扰论的MP2方法[18],选择6-311++g(3df, 3pd)基组,计算驻点的单点能,零点振动能校正后获得驻点的总能量。水液相环境下S-Asp分子的构象1与其前面的2聚水通过氢键作用形成的络合物,记作S-Asp_1·(H2O)2(p),其他体系的标记法相似,反应历程图中键长的单位为纳米(nm)。文中计算由Gaussian09[19]程序完成。
2 结果与讨论
对文献[12]的Asp_1和Asp_2两种构象进行水溶剂化计算,得到它们在水液相下的手性对映体,见图1。计算表明,在碱性水溶液中氢氧根离子水分子簇(OH-·H2O)比它们单独存在具有明显的优势,2聚水(H2O)2也比水分子单独存在具有较大的优势。因此,水液相环境下OH-·H2O和(H2O)2催化Asp异构占主导地位,给予详细讨论,OH-和单个H2O催化Asp异构仅作简要讨论。此外,再对水溶液中的质子在Asp碳负离子的另一侧攻击α-碳,使S-Asp完成手性转变或手性对映体转变做简要讨论。

图1 水液相环境下Asp分子的两种稳定构象的手性对映体Fig.1 Chiral enantiomers of two stable conformations of of Asp molecule in water liquid phase environment
2.1 水液相环境下OH-·H2O催化Asp的手性对映体转变
计算表明Asp_1比Asp_2的构象更稳定,前者的分布具有优势,对其在水液相环境下OH-催化的手性对映体转变机理做详细讨论,对后者仅作一般讨论。
2.1.1 OH-·H2O及(H2O)2催化S-Asp_1手性对映体转变 (OH-·H2O)可以在S-Asp_1的前面与α-氢12H和α-羰基氧9O通过氢键作用形成前驱络合物a_S-Asp_1·(OH-·H2O)(p),也可以与α-氢12H和氨基上的6H通过氢键作用形成络合物b_S-Asp_1·(OH-·H2O)(p),它们均可经过一系列过程实现手性对映体转变,分别称为a通道和b通道。(OH-·H2O)及(H2O)2催化S-Asp_1在a和b通道的手性对映体转变反应历程见图2,反应的势能面见图4。下面分别进行讨论。
1)a通道
第1基元,首先OH-·H2O在S-Asp_1的前面与12H和9O通过氢键作用形成前驱络合物a_S-Asp_1·(OH-·H2O)(p),然后a_S-Asp_1·(OH-·H2O)(p)经H2O辅助OH-抽H的过渡态a_TS1_1·(OH-·H2O)(p),a_S-Asp_1·(OH-·H2O)(p)的α-H解离与OH-形成H2O,异构成中间体络合物a_INT1_1-·(H2O)2(p)(a_INT1_1-的负号表示a_INT1_1是一价碳负离子,下同),接着a_INT1_1-·(H2O)2(p)脱去(H2O)2形成第一中间体碳负离子a_INT1_1-。OH-·H2O与S-Asp_1作用形成a_S-Asp_1·(OH-·H2O)(p)的氢键能是-12.4 kJ/mol,a_INT1_1-·(H2O)2(p)解离成a_INT1_1-和(H2O)2的解离能是37.5 kJ/mol。从a_S-Asp_1·(OH-·H2O)(p)到a_TS1_1·(OH-·H2O)(p),1C—13H键长从0.110 1 nm拉伸至0.139 6 nm,键断裂,骨架二面角5N—1C—3C—8C从126.79°变为136.26°,1个化学键较小的拉伸和较小的骨架形变所需要能量不高,并且a_TS1_1·(OH-·H2O)(p)的键角1C—17H—21O是173.90°,接近平角,过渡态较稳定,因此从a_S-Asp_1·(OH-·H2O)(p)越过a_TS1_1·(OH-·H2O)(p)所需要的能量不高,只有38.7 kJ/mol。
第2基元,碳负离子a_INT1_1-经β-羧基旋转(12C—3C键内旋转)的过渡态a_TS2_1-,异构成第2中间体碳负离子a_INT2_1-。从a_INT1_1-到a_TS2_1-过程,二面角13O—12C—3C—1C从8.38°变为-49.04°,只是二面角旋转所需能量较小,a_TS2_1-产生的能垒只有6.6 kJ/mol。从a_INT1_1-到a_INT2_1-,二面角13O—12C—3C—1C从8.38°变为-86.40°。
第3基元,a_INT2_1-经R-基团旋转(3C—1C键内旋转)的过渡态a_TS3_1-,异构成第3中间体碳负离子a_INT3_1-。从a_INT2_1-到a_TS3_1-,二面角12C—3C—1C—5N从74.17°变为-2.73°,C—C键内旋转所需能量不高,但由于3C—1C键内旋转要带动较大的R-基团旋转较大的二面角,这比前一基元的12C—3C键内旋转只是带动β-羧基旋转需要的能量要多,因此a_TS3_1-产生了14.7 kJ/mol的能垒。从a_INT2_1-到a_INT3_1-,二面角12C—3C—1C—5N从74.17°变为-67.92°,基本实现了二面角12C—3C—1C—5N的对称异构。结构分析表明,从a_INT1_1-到a_INT3_1-,实现了R-基团的对称异构。
第4基元,先是(H2O)2在a_INT3_1-的后面与α-碳1C和α-羧羰基氧9O通过氢键作用形成络合物a_INT3_1-·(H2O)2(q)(q表示(H2O)2在a_INT3_1-的后面,下同),然后经α-碳抽(H2O)2的质子的过渡态a_TS4_1·(OH-·H2O)(q),异构成产物络合物a_P_R-Asp_1·(OH-·H2O)(q)。结构分析表明,a_P_R-Asp_1·(OH-·H2O)(q)与a_S-Asp_1·(OH-·H2O)(p)镜像对称,亦即a_S-Asp_1·(OH-·H2O)(p)实现了手性对映体转变。a_P_R-Asp_1与OH-·H2O的氢键解离后得到S-Asp_1的手性对映体产物a_P_R-Asp_1(与S-Asp_1镜像对称)。a_INT3_1-·(H2O)2(q)的氢键能是-37.5 kJ·mol-1,a_P_R-Asp_1·(OH-·H2O)(q)的解离能是12.4 kJ/mol。从a_INT3_1-·(H2O)2(q)到a_TS4_1·(OH-·H2O)(q),21O—17H键长从0.098 1 nm拉伸至0.122 1 nm,键断裂;活性中心骨架二面角5N—1C—3C—8C从-167.55°变为-136.26°,1个化学键的拉伸和骨架形变所需要能量不太高。又a_TS4_1·(OH-·H2O)(q)的键角1C—17H—21O是173.90°,接近平角,过渡态较稳定;从a_INT3_1-·(H2O)2(q)到a_TS4_1·(OH-·H2O)(q),α-碳从sp2杂化变为准sp3杂化,从非满配状态向准满配状态过渡是放热过程,又O—H键断裂比C—H键断裂消耗的能量小得多,因此a_TS4_1·(OH-·H2O)(q)产生的能垒要小于a_TS1_1·(OH-·H2O)(p)产生的能垒许多,只有14.3 kJ/mol。
2)b通道
第1基元,首先OH-·H2O在S-Asp_1前面与12H和6H通过氢键作用形成络合物b_S-Asp_1·(OH-·H2O)(p),然后b_S-Asp_1·(OH-·H2O)(p)经水分子辅助OH-抽氢的过渡态b_TS1_1·(OH-·H2O)(p),α-H与OH-形成水分子,异构成中间体碳负离子络合物b_INT1_1-·(H2O)2(p),接着b_INT1_1-·(H2O)2(p)脱去(H2O)2形成第一中间体碳负离子b_INT1_1-。b_S-Asp_1·(OH-·H2O)(p)的氢键能是-11.6 kJ·mol-1,b_INT1_1-·(H2O)2(p)解离成b_INT1_1-和(H2O)2的解离能是37.8 kJ/mol。从b_S-Asp_1·(OH-·H2O)(p)到b_TS1_1·(OH-·H2O)(p),1C—21H键长从0.109 6 nm拉伸至0.140 0 nm,键断裂,活性中心骨架二面角5N—1C—3C—8C从130.83°变为139.70°,1个化学键较小的拉伸和较小的骨架形变所需要能量不高,又b_TS1_1·(OH-·H2O)(p)的键角1C—21H—17O是172.67°,接近平角,过渡态较稳定,因此b_TS1_1·(OH-·H2O)(p)产生的能垒只有39.4 kJ/mol。
结构分析表明,b_INT1_1-全同于a_INT1_1-。b_INT1_1-经过与a通道第2和第3基元相同的历程异构成b_INT3_1-(a_INT3_1-)。b_INT3_1-后面的(H2O)2与其1C和7H氢键作用形成b_INT3_1-·(H2O)2(q),经与a_TS4_1·(OH-·H2O)(q)相似的过渡态异构成与b_S-Asp_1·(OH-·H2O)(p)镜像对称的络合物b_R-Asp_1·(OH-·H2O)(p),S-Asp_1在b通道实现手性对映体转变,详细机理从略。

图2 水液相环境下氢氧根水分子簇催化S-Asp_1手性对映体转变的反应历程Fig.2 Reaction process of chiral enantiomer transition of S-Asp_1 catalyzed by hydroxyl ion water clusters in water liquid phase environment
2.1.2 OH-·H2O催化S-Asp_2手性对映体转变的决速步骤 研究表明,S-Asp_2的手性对映体转变历程相似于S-Asp_1的手性对映体转变历程,决速步骤也是第1基元反应,为节省篇幅,这里仅对决速步骤并且是OH-·H2O在S-Asp_2的前面与α-氢16H和α-羧羰基氧9O通过氢键作用形成络合物的情况进行讨论。
S-Asp_2的手性对映体转变决速步骤的反应历程,见图3,反应的势能面见图4。
首先OH-·H2O在S-Asp_2的前面与16H和9O通过氢键作用形成络合物S-Asp_2·(OH-·H2O)(p),此过程放热14.0 kJ/mol。然后S-Asp_2·(OH-·H2O)(p)经过渡态TS1_2·(OH-·H2O)(p),异构成中间体碳负离子络合物INT1_2-·(H2O)2(p),此过程OH-抽取了S-Asp_2的α-H形成H2O,S-Asp_2变成了天冬氨酸碳负离子INT1_2-。从S-Asp_2·(OH-·H2O)(p)到TS1_2·(OH-·H2O)(p),1C—16H键长从0.109 3 nm拉伸至0.138 9 nm键断裂,5N—1C键长从0.145 7 nm拉伸至0.146 6 nm;骨架二面角5N—1C—3C—8C从125.39°变为135.99°;二面角9O—8C—1C—5N从-12.18°变为21.10°,α-羧基旋转了33.28°。1个化学键的断裂、1个化学键的拉伸、骨架形变及羧基旋转所需能量会高与前面的S-Asp_1·(OH-·H2O)(p)到TS1_1·(OH-·H2O)(p)过程。但由于TS1_2·(OH-·H2O)(p)的氢键角1C—19H—19O是172.50°,过渡态较稳定,因此从S-Asp_2·(OH-·H2O)(p)越过TS1_2·(OH-·H2O)(p)所需要的能量又不会太高,只有40.2 kJ/mol。最后,INT1_2-·(H2O)2(p)脱去(H2O)2形成第一中间体碳负离子a_INT1_2-,解离能是37.2 kJ/mol。

图3 氢氧根水分子簇催化S-Asp_2手性转变决速步骤的反应历程Fig.3 Reaction process of the rate-determining step of chiral transition of S-Asp_2 catalyzed by hydroxyl ion water clusters

图4 水液相环境下氢氧根水分子簇催化S-Asp手性对映体转变反应的势能面Fig.4 Potential energy surfaces of chiral enantiomer transition of S-Asp catalyzed by hydroxyl ion water clusters in water liquid phase environment
单从图4看,S-Asp手性对映体转变反应的决速步视乎应该是第一基元反应产物的解离过程,计算表明,a_INT1_1-·(H2O)2(p)、b_INT1_1-·(H2O)2(p)和INT1_2-·(H2O)2(p)可以直接进行与图4的第2和第3基元相似的过程异构,反应能垒也基本同于图4的第2和第3基元,实际的反应应该是这样的,如此讨论只是为了使问题简化而已;图4的第4基元实际也可以是a_INT3_1-·(H2O)2(p)与其后面的(H2O)2络合形成a_INT3_1-·(H2O)2(p)·(H2O)2(q),经和第4基元相似的过渡态异构成产物络合物a_P_R-Asp_1·(H2O)2(p)·(OH-·H2O)(q),能垒也是基本不变。因此,在考察哪一步是决速步时无需考虑络合物的氢键解离过程。由于本工作研究的是水溶剂里的反应,形成氢键所放热量会迅速地被水吸收,不会为接下来的反应提供能量,反应活化能应按内禀能垒计算。因此,Asp_1和Asp_2经图4的4个基元反应实现手性对映体转变时,决速步能垒应是第一基元反应的前驱络合物越过过渡态所需的能量。OH-·H2O抽氢Asp_1在a和b通道手性对映体转变决速步的内禀能垒分别是34.1和34.7 kJ/mol,在误差允许的范围内可认为相等,大约是34.0 kJ/mol左右;OH-·H2O抽氢,Asp_2手性对映体转变决速步的内禀能垒是40.2 kJ/mol。这几个质子迁移反应的能垒均小于温和反应的能垒80.1 kJ/mol[20]许多,说明水溶剂环境下OH-·H2O的催化可以使Asp的手性对映体转变顺利进行,亦即实验上可以在碱的水溶液环境下实现Asp的手性对映体转变,同时也说明生命体内过多的OH-会严重影响健康。水溶剂环境下OH-·H2O抽氢Asp手性对映体转变决速步的内禀能垒与文献[12]的(H2O)2催化Asp手性对映体转变的决速步能垒(约106.0 kJ/mol)相比较大幅降低,说明OH-·H2O对Asp手性转变反应的催化作用远远好于(H2O)2。
2.2 水液相环境下氢氧根催化S-Asp手性对映体转变
同于前面的讨论,OH-抽取α-H的反应也是OH-催化S-Asp手性对映体转变的决速步,OH-抽取α-H后Asp碳负离子继续异构的过程同于OH-·H2O取α-H后Asp碳负离子的异构,因此这里只讨论OH-抽取α-H的过程,为使问题简便,S-Asp与OH-及Asp碳负离子与H2O分子之间氢键的形成与解离过程不给予讨论。
2.2.1 水液相环境下氢氧根催化S-Asp_1手性对映体转变的决速步骤 水液相环境下氢氧根催化S-Asp_1手性对映体转变决速步骤的反应历程,见图5,反应的势能面见图7的第1个图。
S-Asp_1·OH-(p)经OH-抽氢的过渡态TS1_1·OH-(p),α-H与OH-形成水分子,异构成中间体络合物INT1_1-H2O(p)。从S-Asp_1·OH-(p)到TS1_1·OH-(p),1C—12H键长从0.109 4 nm拉伸至0.137 6 nm,伸长0.028 2 nm,键断裂,拉伸幅度小于a_S-Asp_1·(OH-·H2O)(p)到a_TS1_1·(OH-·H2O)(p)的0.029 5 nm;骨架二面角5N—1C—3C—8C从126.48°变为135.31°,形变8.83°,活性中心骨架形变小于从a_S-Asp_1·(OH-·H2O)(p)到a_TS1_1·(OH-·H2O)(p)过程的9.47°。因此TS1_1·OH-(p)产生的能垒只有27.6 kJ/mol,小于a_TS1_1·(OH-·H2O)(p)产生的能垒。

图5 水液相环境下氢氧根催化S-Asp_1手性转变决速步骤的反应历程Fig.5 Reaction process of the rate-determining step of chiral transition of S-Asp_1 catalyzed by hydroxyl ion in water liquid phase environment
2.2.2 水液相环境下OH-催化S-Asp_2手性对映体转变的决速步骤 水液相环境下OH-催化S-Asp_2手性对映体转变决速步骤的反应历程,见图6,反应的势能面见图7的第2个图。
S-Asp_2·OH-(p)经OH-抽氢的过渡态TS1_2·OH-(p),α-H与OH-形成水分子,异构成中间体络合物INT1_2-·H2O(p)。从S-Asp_2·OH-(p)到TS1_2·OH-(p),1C—12H键长从0.109 4 nm拉伸至0.136 8 nm,伸长了0.027 4 nm键断裂,拉伸幅度小于从S-Asp_2·(OH-·H2O)(p)到TS1_2·(OH-·H2O)(p)过程的0.029 6 nm;骨架二面角5N—1C—3C—8C从125.17°变为134.56°,形变9.39°,骨架形变小于从S-Asp_2·(OH-·H2O)(p)到TS1_2·(OH-·H2O)(p)过程的10.60°,且从S-Asp_2·OH-(p)到TS1_2·OH-(p)过程α-羧基基本无旋转{从S-Asp_2·(OH-·H2O)(p)到TS1_2·(OH-·H2O)(p)过程α-羧基旋转明显}。因此,TS1_2·OH-(p)产生的能垒小于TS1_2·(OH-·H2O)(p)的能垒,只有26.8 kJ/mol。
从图7可以看出,OH-抽α-氢致Asp_1和Asp_2手性转变的决速步内禀能垒分别是27.6和26.8 kJ/mol。这两个质子迁移反应的能垒均远小于温和反应的能垒80.1 kJ/mol[20],说明水溶剂环境下OH-的催化可以使Asp的手性对映体转变反应进行的较快。同图4比较可知,OH-抽α-H致Asp_1和Asp_2手性转变比OH-·H2O抽α-H在热力学上具有优势。同2.1一样本节的工作也说明实验上可以在碱的水溶液环境下实现Asp的手性对映体转变,从理论上对文献[16]的实验研究给予了合理的解释,同时也说明生命体内过多的OH-会严重影响健康。
2.3 质子攻击Asp碳负离子的α-碳形成R-Asp的机理
a_INT1_1-里侧的空间狭小,由于空间位阻效应,水分子(簇)很难在纸面里侧与a_INT1-_1通过氢键作用形成络合物,因此,a_INT1-_1的α-碳很难抽取水分子中的质子形成a_R-INT1_1(是S-Asp_1旋光异构的产物之一,但不是手性对映体转变)。带正电荷且线度较小的质子攻击碳负离子时没有这些困难,质子攻击碳负离子a_INT3_1-时可以得到S-Asp_1的手性对映体产物a_P_R-Asp_1,因此为节省篇幅又不失问题讨论的一般性,仅对质子在纸面里侧攻击S-Asp_1手性对映体转变a路径上的中间体碳负离子a_INT1_1-和a_INT3_1-的情况进行讨论。
质子攻击a_INT1_1-和a_INT3_1-形成a_R-INT1_1和a_P_R-Asp_1的简明过程见图8的
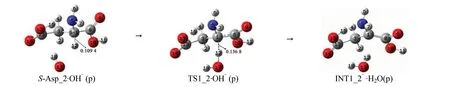
图6 水液相环境下氢氧根催化S-Asp_2手性转变决速步骤的反应历程Fig.6 Reaction process of the rate-determining step of chiral transition of S-Asp_2 catalyzed by hydroxyl ion in water liquid phase environment

图7 水液相环境下氢氧根催化S-Asp手性对映体转变反应决速步骤的势能面Fig.7 Potential energy surfaces of chiral enantiomer transition of S-Asp catalyzed by hydroxyl ion in water liquid phase environment
(A)与(B)(其中a_R-INT1_1和a_P_R-Asp_1是对质子攻击到势能最低点时的驻点优化后的构型),这两个过程刚性扫描的势能曲线见图9的(A)与(B)。
从势能曲线可以看出,质子与a_INT1_1-的1C距离大于0.475 0 nm时,质子与a_INT1_1-的相互作用几乎不存在,它们的距离为0.109 5 nm时,相互作用最强,形成a_R-INT1_1,可以看成是S-Asp_1手性对映体转变过程中的旋光异构产物(只是手性转变,非手性对映体转变)。
质子与a_INT3_1-的1C距离大于0.500 0 nm时,质子与a_INT3_1-的相互作用几乎不存在,它们的距离为0.109 4 nm时,相互作用最强,形成a_P_R-Asp_1,亦即S-Asp_1实现了手性对映体转变。

图8 H+攻击a_INT1-_1和a_INT3_1-的α-C形成a_R-INT1_1和a_P_R-Asp_1的过程Fig.8 Process of protons attack on α-carbon that a_INT1_1- and a_INT3_1- form a_R-INT1_1 and a_P_R-Asp_1

图9 H+攻击a_INT1_1-和a_INT3_1-的α-C过程的势能曲线Fig.9 Potential energy curve of protons attack on α-carbon of INT1_1 and a_INT3_1-
图9说明,质子攻击a_INT1_1-和a_INT3_1-形成a_R-INT1_1和a_P_R-Asp_1的过程均是无势垒放热过程,溶液中质子的存在会使S-Asp手性转变反应的后半程迅速进行。图8及9说明,质子攻击a_INT1-_1的α-碳空间位阻不复存在,质子在纸面里攻击中间体碳负离子的α-碳可以得到S-Asp不同的消旋体产物(含非手性对映体产物)。图9还说明,在势能极小值处质子继续攻击α-碳是强烈的吸热过程,产物越来越不稳定,当质子与α-碳的距离趋于零时产物能量趋于无穷大,亦即要想让质子进入S-Asp的α-碳几乎是不可能的。
3 结 论
1)Asp分子经过氢氧根离子(水分子簇)和碳负离子的α-碳抽氢及α-羧羟基、β-羧羟基、β-羧基和R-基旋转的一系列过渡态,可以实现手性对映体转变。质子攻击S-Asp手性转变反应的中间体碳负离子会加快S-Asp手性转变的进程,同时还会得到与S-Asp非镜像对称的旋光异构体。
2)OH-抽α-氢是Asp_1和Asp_2旋光异构的决速步骤,只是OH-抽α-氢致Asp_1和Asp_2旋光异构的决速步内禀能垒在27.0 kJ/mol左右;水分子辅助OH-抽α-氢致Asp_1和Asp_2旋光异构的决速步内禀能垒分别是34.0和40.0 kJ/mol。OH-抽α-氢致Asp_1和Asp_2手性转变比氢氧根水分子簇抽α-氢在热力学上具有优势。
3)质子攻击碳负离子的α-碳形成稳定产物的过程是无势垒放热过程,溶液中质子的存在会加快S-Asp手性转变后半程反应的速度。
结果表明,生命体内OH-的存在是S-Asp消旋的原因,质子的存在又加快了这一过程,这指导人们要均衡地的摄入碱性与酸性食物;在实验上可以在碱性环境下将Asp的所谓劣构体转变成为优构体。
