使用R 分析单核苷酸多态性位点-环境交互作用 对疾病影响的方法
2020-02-23张漪丽
张漪丽,陈 昕
(湖南师范大学医学院,长沙 410013)
R 语言是由新西兰奥克兰大学的罗斯·伊哈卡和罗伯特·杰特曼开发的一款免费开源统计软件。相较于其它商业统计软件,R 可以根据自身需求创建自定义函数并以包(packages)的形式分享给世界上各地的使用者,达到各种功能的即时装载和演算。目前,R 在世界上已经拥有了近千万的用户,在流行病学、统计学、生命科学、金融学、农学等领域广泛应用[1]。单核苷酸多态性(Single nucleotide polymorphisms,SNPs)指由单个核苷酸的改变而引起的DNA 序列的改变,造成包括人类在内的物种之间染色体基因组的多样性。自从2008 年全基因组关联研究的方案提出至今,人类已经建立了完备的基因数据库,大量SNPs 位点被测序研究。[2]目前已经证实,SNPs 位点的突变与疾病发生发展呈高度关联,研究SNPs 位点对疾病的作用,继而以此作为有效的筛检指标在早期预防疾病已成为流行病学研究的热点。并且在生物医学和临床医学领域,研究个案的SNPs 位点对个体化医疗和精准医疗提供了理论支撑。交互作用是在各学科研究中常见的问题。指纳入研究的两个以上的因素之间产生相互效应,使因素联合作用时产生的效应不等于单独使用时的效应之和[3]。交互作用按照方向分为协同作用和拮抗作用,按照意义分为统计学交互作用、生物学交互作用和公共卫生学交互作用。在流行病学和生物医学的研究中,常通过统计学交互作用为探讨生物学交互作用提供线索[4]。
1 材料与方法
1.1 材料 本研究所使用的资料为R 包“SNPassoc”中内置数据集“SNPs”,调用ID 号,病例/对照分组,性别和35 个SNP 位点,共157 个案例。建立虚拟因子“risk”作为环境危险因素纳入数据集,“risk”的取值为二分类0,1变量,其值通过设置种子数,用sample 函数随机产生,个数与案例数相同。以此演示两因素的交互作用分析。
1.2 方法 使用自建函数将单水平的SNP 位点筛除。SNPs 位点通过“SNPassoc”包进行模型筛选。模型建立Logistic 回归模型,调整变量为性别。显性模型、隐性模型、超显性模型作为二分类变量纳入研究,共显性模型作为三分类变量纳入研究。最优模型选择根据赤池信息标准值(Akaike information criterion,AIC)界定。分别建立两个Logistic 回归模型,因变量为是否患病,自变量为risk 和SNPs 候选位点,第一个模型仅考虑risk 和SNPs的独立作用,第二个模型在此基础上加入两个因素的交互项,全部建立新的哑变量后再纳入研究,其中risk 0 值为未暴露,1 为有暴露。似然比检验被用于两个Logistic回归模型的统计学分析,似然比计算为卡方检验法,当P 值显著说明交互项在模型间有统计学意义。使用R“epiR”包对显著交互作用的SNP 位点进行定量分析。使用超频相对危险度(relative excess risk of interaction,RERI)和协同指数(synergy index,S)指标进行评价。本研究中所有分析P<0.05 被认为显著。所有统计学分析均在R 软件(https://www.r-project.org/,版本:3.6.2)上完成。
2 结果
2.1 数据集建立 根据内置数据集建立本研究的演示集,建立集的代码如下:
#建立数据集
library("SNPassoc")
data(SNPs)
SNPs<-SNPs[,-c(4,5)]
set.seed(2020)
risk<-sample(0:1,157,replace=T)
SNPs<-cbind(risk,SNPs)
2.2 SNPs 筛选和模型选择 将候选35 个SNPs 位点中仅有一个水平的位点剔除,之后将符合纳入标准的位点通过“SNPassoc”包建立调整Logistic 回归模型,将SNPs 变换为最优模型纳入后续研究。最后有21 个位点纳入后续研究。图1 展示了数据集中所有SNPs 位点的显著性。表1 展示了SNPs 位点模型的选择结果和AIC 检验值。
2.3 SNPs-环境危险因素交互作用检验 建立条件Logistic 回归模型和似然比检验对SNP-环境危险因素进行交互作用检验。经检验,共有1 个SNP 位点snp10008 显著,表2 展示了显著位点信息和似然比检验结果。可知snp10008(隐性模型 OR=1.51,95%CI=0.30-7.56)和危险因素risk 在疾病中存在交互作用(似然χ2=4.11,P=0.043)。
2.4 计算交互作用指标 表3 展示了snp10008 位点的Logistic 检验结果,使用“epiR”包进行交互作用计算。RERI=-0.0036(95%CI=-0.236-0.2358),S=0.244(95%CI=0.132-0.541)。交互作用的解释受限。
3 讨论
本文通过分析程序包内置的SNPs 位点的模拟数据集,演示了如何对SNPs-环境的交互作用进行统计学分析和解释。由于资料并不是真实生物医学研究中的SNP,部分位点有逻辑错误,并没有研究的意义。并且由于157 的样本量相较于真实研究中过少,部分位点呈单水平或者不通过哈温平衡检验,损失了部分信息。
本研究中评价交互作用的方法为似然比检验法,在SNP 相关研究中,相较于Wald 系数检验法,似然比检验由于其特征,规避了可能的混杂效应,更加精确可靠,受到广泛的认可[5]。

表1 候选SNPs位点信息及模型选择
根据国内外学者的研究报告,在生物学交互作用的研究中,相加模型的统计指标相较于相乘模型更能反映真实的效应[6]。然而,本研究选择Logistic 回归法,本质仍是相乘模型。原因是目前在研究SNPs 交互作用中,Logistic 回归仍为最广泛而有效的手段。为了探索生物学意义,我们在相乘模型的结果上纳入了RERI 和S 等相加模型指标进行分析,以期综合评价。值得注意的是,相乘模型指标的比值大于1 时,相加模型检验指标也有协同的显著性意义。但是相乘模型小于1 时,相加模型检验指标会出现协同、拮抗、无统计学意义三种可能情况[7]。其原因是统计学的交互作用与生物学的交互作用是复杂的,并没有绝对的对应关系。因此,在遇到这种情况时,应该结合自身的专业知识和既往研究报告,做出合理的判断。比如在SNP 研究中可以查询该位点的文献报告情况和位置基因,判断基因是否有与待研究因素的特征。

图1 候选SNPs位点模型显著性分析

表2 Snp10008位点的模型特征与似然比检验结果
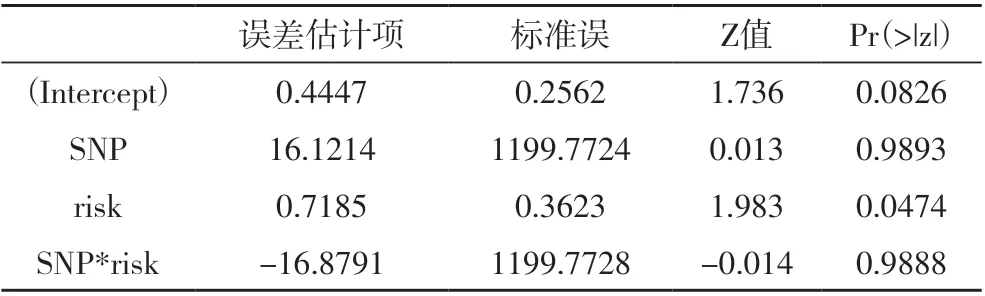
表3 Logistic回归分析结果
本研究中的显著位点snp10008 由于样本量有限导致其在计算参数时标准误过大,在研究中通常要考虑采用其他指标评估是否SNP 位点真正显著,而不能仅考虑logistic 回归值。由于数据不够理想,未显示出交互作用。 另外,本文使用的是参数检验法,研究SNPs 交互作用还有一种非参数检验法,广义多因子降维法,该方法采用数学模型对研究因素进行降维,弥补了Logistic回归在处理高阶交互作用时的局限性[8]。在常见的慢性病如高血压糖尿病中已有广泛而成功应用。但是值得注意的是,GMDR 对数据要求较高,并且采用无模型方法,不可完全替代传统回归分析法。
随着计算机技术和人工智能的发展,高通量基因组学的研究成为现实,SNPs 位点已经成为生物医学研究的热点。而R 语言拥有良好的生态系统,软件包多为专业的统计学家或计算机专业人员制作,并在公布前进行内部同行评审,保证了该统计软件的精确性和可靠性。本文旨在提供新的研究方法,其中使用到的所有R 包已在基因组学研究中广泛应用,为流行病学研究人员带来了便利。


