灵芝多糖的结构及其表征方法研究进展
2020-01-16聂少平艾连中夏永军熊智强王光强
张 汇 聂少平 艾连中 夏永军 熊智强 王光强*
(1 上海理工大学医疗器械与食品学院 上海食品微生物工程技术研究中心 上海 200093
2 南昌大学食品科学与技术国家重点实验室 南昌 330047)
灵芝属真菌门、担子菌纲、多孔菌科、灵芝科、灵芝属,主要生长于朽木或树桩上,在我国及亚洲几个国家(如日本、韩国)广泛使用,被认为具有调节人体机能平衡和延年益寿的功效[1]。灵芝种类繁多,目前全球已发现并报道的灵芝品种超过80个,然而,由于灵芝同义词的滥用,所以很难对灵芝进行精确的种属划分[2]。日本的“Reishi”,韩国的“Youngzhi”以及中国的“灵芝”,在英文中都被翻译成“Ganoderma”,它们是否属于同一实体,还有待商榷。在我国古典药书《神农本草经》中,灵芝按颜色划分为青、赤、黄、白、黑和紫6种;此外,我国还分布有松杉灵芝、树舌、薄盖灵芝、云芝等。
灵芝子实体中的化学成分丰富,被认为是活性物质的生物制造工厂[3]。多糖和三萜类物质是其中最主要的功能成分,此外,还含有核酸类、呋喃类、甾醇类、生物碱类和氨基酸多肽类等[4]。自1981年 Miyazaki等[5]从灵芝子实体(G.lucidum)中分离出高支化的阿拉伯木糖基葡聚糖,并证实具有抗肿瘤活性以来,关于灵芝多糖的报道越来越多。大部分报道的灵芝多糖为结构复杂的杂多糖,主要由葡萄糖、半乳糖、甘露糖、岩藻糖、木糖和阿拉伯糖通过不同比例和不同糖苷键类型连接构成,部分灵芝多糖上还复合了蛋白或多肽残基以及酚类物质等。而对于灵芝多糖的功能研究则主要集中于生物活性功能,现有研究表明:灵芝多糖在免疫调节,抗肿瘤,抗氧化,降血糖,调节肠道健康等方面具有良好的生理功能[3,6],由此也引起了国内外学者的广泛关注。众所周知,灵芝多糖的生物活性与其化学结构和构象特征有着紧密的联系。要研究灵芝多糖生物活性的机理及其构效关系,就需要对其结构特征进行详细、准确的表征。为此,本文总结近年来灵芝多糖的结构特征研究情况,在此基础上,结合现代仪器分析技术,综述多糖结构表征的方法学研究进展,为灵芝多糖一级结构和高级结构的深入研究提供参考。
1 灵芝多糖的提取与分级
根据多糖在灵芝中的存在部位及其溶解性,可分别采用热水浸提[7]、盐溶液浸提[8]和碱液提取[9]等方式获得灵芝多糖。热水和盐溶液提取法遵循中药的煎煮原则,具有简便安全和环保等特点,是最常见的多糖提取方法;而碱液提取主要是从细胞壁中萃取出不溶于水的高分子质量的多聚糖。不同溶剂提取得到的灵芝多糖结构存在较大差异,通常,热水提取的多糖为水溶性杂多糖,而碱液提取的多糖为水不溶性的葡聚糖。
多糖的分离与分级方法包括Sevage法脱蛋白、乙醇分级沉淀法、季铵盐沉淀法、冻融法、阴离子交换柱层析、凝胶渗透色谱、膜分离法等[10]。分级方法的选择取决于多糖的性质、组成及分离的目的等,总体遵循先除杂,后细分的原则。灵芝子实体的水提物是一种非常复杂的混合物,其中含有一定量的色素类物质,因此需要对其进行脱色处理。常用的脱色方法包括双氧水氧化法和活性炭吸附法,然而,这两种方法都会影响多糖的结构或得率[11]。目前,阴离子交换色谱被广泛用于多糖的脱色和分级[12]。表1总结了不同分级方法的原理及其在灵芝多糖中的应用。

表1 多糖分级的方法及其在灵芝多糖中的应用Table1 Methods for fractionation of polysaccharides and the application on Ganoderma polysaccharides
在灵芝多糖的分级过程中,通常需要结合2~3种分级方法,才能达到对样品的纯度要求,其中,阴离子交换色谱结合凝胶渗透色谱是最常见的分级手段[16-18],其主要原理是首先通过阴离子交换色谱将灵芝多糖中的中性多糖和酸性多糖进行分离,再通过凝胶渗透色谱对不同分子质量的多糖级分进行分级。Zhang等[21]也报道直接采用阴离子交换色谱可将灵芝多糖中不同性质的多糖成分和杂质进行分离,得到高纯度的多糖组分。
2 灵芝多糖的一级结构
2.1 灵芝多糖的单糖组成
多糖的一级结构包括单糖组成、糖连接方式和连接序列等。不同来源的灵芝多糖其单糖组成虽存在差异性,但主要以葡萄糖和半乳糖为主,有些还含有甘露糖、岩藻糖、木糖和阿拉伯糖等。Ye等[22]从赤灵芝子实体中分离得到了一种岩藻糖基半乳聚糖,通过高效阴离子交换色谱-脉冲安培检测技术(HPAEC-PAD)证实其由岩藻糖、葡萄糖和半乳糖以1∶1∶5的比例组成;Wang等[16]从松杉灵芝中分离得到了14种水溶性和15种水不溶性的多糖组分,发现水溶性的多糖为结合有蛋白的葡萄糖基半乳聚糖,单糖组成包括葡萄糖、半乳糖、岩藻糖和甘露糖,而水不溶性的多糖为结合有蛋白的β-(1→3)-葡聚糖;从黑灵芝不同部位(菌柄、菌盖和孢子粉)中提取得到的多糖,其单糖均主要由葡萄糖、半乳糖和甘露糖组成,而比例有显著差别,其中,黑灵芝孢子粉多糖主要由葡萄糖组成,含有极少量的半乳糖和甘露糖,而菌柄和菌盖多糖中半乳糖和甘露糖的含量则有所提高[23]。
2.2 灵芝多糖的化学结构
根据现有研究报道,灵芝多糖按化学组成可分为均多糖和杂多糖两大类,其中均多糖主要为葡聚糖类;而杂多糖的结构则较为复杂。
β-(1→3)-葡聚糖被认为是具有良好生物活性的多糖化合物,可是由于其可形成刚性的螺旋结构,在水中的溶解性很差,需要用碱性溶液进行溶解。Ukai等[24]首次从紫灵芝子实体中分离得到了一种碱溶性的β-(1→3)-葡聚糖;随后,Sone等[25]从赤灵芝子实体和菌丝体培养液中提取得到了水不溶性的β-(1→3)-葡聚糖。水不溶性的β-(1→3)-葡聚糖为支化度较低的线型分子,呈刚性链状;而对含有支链的的葡聚糖,其水溶性可大大改善。Liu等[13]采用热水浸提从赤灵芝子实体中分离出一种高分子质量的β-葡聚糖,证实其分子结构单元是以β-(1→3)-葡聚糖为主链,每隔两个葡萄糖中有一个O-6位的支链(图1)。Bao等[18]从赤灵芝子实体中分离得到一种水溶性的高支化葡聚糖,其中主链由β-(1→3)-葡萄糖构成,而支链点出现在O-6位置,支链结构为(1→6)-葡萄糖。Amaral等[26]则从无柄灵芝(G.resinaceum)中分离得到了一种高支化的水溶性β-(1→3)-葡聚糖,在O-6位置出现支链点,支链结构为(1→4)-葡萄糖。此外,灵芝孢子粉中也含有大量的β-(1→3)-葡聚糖,Bao等[27]报道了赤灵芝孢子粉中的主要多糖为以β-(1→3)-葡萄糖为主链,支链为不同聚合度的β-(1→3)和(1→6)-葡萄糖链,刚果红试验证明该多糖可形成与香菇多糖类似的三股螺旋结构,具有良好的免疫调节功能。

图1 赤灵芝子实体中β-葡聚糖的化学结构单元[13] Fig.1 Repeating unit ofβ-Glucan from the fruiting bodies of Ganoderma lucidum[13]
灵芝中还含有一类葡聚糖:α-(1→3)-葡聚糖,它主要存在于细胞壁内,因此很难直接用热水提取出来,而需要用盐溶液或碱液进行萃取,分离得到的α-(1→3)-葡聚糖通常为水不溶性的多糖。Bao等[28]采用碱液从赤灵芝的破壁孢子粉中分离得到一种水不溶性多糖,通过核磁共振(NMR)和红外光谱(FT-IR)证实其为线型的α-(1→3)-葡聚糖,通过化学改性制备了该多糖的不同修饰产物,结果表明水不溶性的α-(1→3)-葡聚糖没有生物活性,而修饰产物的水溶解性大大改善,并具有良好的免疫调节活性;Zhang等[9]通过碱液提取从黑灵芝子实体中分离得到一种高分子链的线型α-(1→3)-葡聚糖,该多糖不溶于水,而通过硫酸化修饰可有效改善其水溶解性,且修饰产物具有良好的抑制肿瘤细胞生长的作用。
灵芝中的杂多糖种类很多,主链主要以葡萄糖、半乳糖和甘露糖为主,支链中含岩藻糖、木糖和阿拉伯糖等糖单元。Miyazaki等[5]首次报道的赤灵芝多糖是一种水溶性的高支化阿拉伯木糖基葡聚糖,主链主要由 α和β-(1→4)-葡萄糖、β-(1→6)-和β-(1→3)-葡萄糖构成。而 Usui等[20]则首次从树舌灵芝中分离得到两种杂半乳聚糖,其主链均由 α-(1→6)-半乳糖构成,且均被 O-乙酰基部分取代,支链中则由岩藻糖和甘露糖组成。Bao等[18]从赤灵芝子实体中分离得到两种杂多糖(PL-1和PL-4),其主链分别由 α-D-(1→4)-葡萄糖、β-D-(1→6)-半乳糖和β-(1→3),β-(1→4),β-(1→6)-葡萄糖、β-(1→6)-甘露糖组成。Ye等[29]则从赤灵芝子实体中分离得到了一种糖肽,其聚糖部分的主链由 α-(1→6)-半乳糖和α-(1→3)-葡萄糖构成,支链点出现在半乳糖的O-2位置,支链则由岩藻糖组成。Zhang等[30-31]从黑灵芝中分离得到了两种低分子量的水溶性杂多糖PSG-1-F0.2和PSG-2,其中,PSG-1-F0.2是一种高支化的酸性 β-(1→3,1→6)-葡聚糖,其化学结构的重复单元如图2a所示;而PSG-2则为一种含有乙酰基的杂半乳聚糖,其主链为α-(1→6)-半乳聚糖,支链中含有岩藻糖、甘露糖和葡萄糖,其重复单元结构如图2b所示。

图2 黑灵芝子实体中两种水溶性多糖的重复单元结构[30-31] Fig.2 Repeat unit of two water-soluble polysaccharides from the fruiting bodies of Ganodernra atrum[30-31]
综上可知,灵芝多糖种类繁多,结构复杂,有报道的灵芝多糖结构已达200多种,其中既含有杂多糖,也包含葡聚糖;其单糖主要由葡萄糖、半乳糖、甘露糖、木糖和岩藻糖等组成,而单糖间的糖苷键包括(1→3)、(1→4)、(1→6)等连接方式;大多数灵芝多糖有分支,部分还含有蛋白或多肽片段,形成糖蛋白或蛋白聚糖结构[32]。
3 灵芝多糖的分子质量及其构象特征
不同种类或不同来源的灵芝,其多糖成分的分子质量(Mw)有较大差异,主要在104u数量级,也有报道灵芝多糖的Mw在106u范围。灵芝β-葡聚糖是灵芝中的主要活性成分,其分子质量分布在104~105范围,Bao等[18]报道的赤灵芝子实体中β-(1→3)-葡聚糖的Mw为6.3×104u;无柄灵芝中的高支化 β-(1→3)-葡聚糖的Mw为2.6×104u[26];而 Liu 等[13]报道的赤灵芝子实体中 β-(1→3)-葡聚糖则为高分子质量的级分,其在水溶液中的Mw为3.75×106u。灵芝 α-(1→3)-葡聚糖的Mw 则相对较大,分布在105~106u范围内,如赤灵芝孢子粉中的α-(1→3)-葡聚糖的Mw为1.26×105u[28],而黑灵芝中 α-(1→3)-葡聚糖的Mw则为1.665×106u[9]。灵芝中的杂多糖多为水溶性较好的多聚糖,其分子质量相对较小,主要分布在103~104u范围内,如赤灵芝中的阿拉伯木糖基葡聚糖的Mw为4×104u[32],松杉灵芝中的β-半乳糖基-α-甘露聚糖的Mw为8.35×104u[33],而从黑灵芝中分离得到的一种杂半乳聚糖Mw为6.9×104u[31];此外,Huang等[34]报道了一种高分子质量的赤灵芝中性多糖,其 Mw 达 2.5×106u。
构象属于多糖的高级结构,研究表明,多糖的功能性质不仅与其化学结构相关,还与其在溶液中的空间构象有着密切联系[35]。多糖在溶液中的构象特征表征方式有多种,如多糖分子的尺寸、大小、形态、黏度、柔顺性/刚性等,多糖在溶液中的形态主要包括:球形链、无规线团、半柔性链、刚性棒状、单股螺旋、双股螺旋和三股螺旋等[36]。
灵芝中的β-(1→3)-葡聚糖由于其结构较为规整,容易形成螺旋结构,因此,其在溶液中通常呈现为刚性链状。Bao等[27]通过刚果红实验证实从赤灵芝孢子粉中提取得到的葡聚糖SP在水溶液中的构象与Curdlan的三股螺旋构象相似,从而证明SP在水溶液中呈现一定的有序构象。从赤灵芝子实体中分离得到的一种高分子质量的β-(1→3)-葡聚糖,光散射技术研究表明该多糖在0.9%的NaCl溶液中呈刚性链状,而在DMSO中则呈无规线团[13]。
对于支化度较小的灵芝杂多糖,由于结构较为复杂、糖苷键连接方式多样,其在溶液中通常呈现为无规线团或棒状结构;而对于高支化的灵芝多糖,则呈现紧凑卷曲的结构。Peng等[37]对松杉灵芝菌丝体中提取得到的一种碱溶性多糖GM5-1的链构象进行研究,测定了GM5-1在0.25 mol/L LiCl/DMSO溶液中的Mw及特性黏度[η],并建立了[η]与Mw以及回旋半径Rg与Mw之间的关系,结果表明GM5-1在0.25mol/L LiCl/DMSO溶液中是以半柔性链的构象存在。同时,Peng等[38]对松杉灵芝菌丝体中提取得到的另一种碱溶性多糖GM6-1的溶液性质也进行了研究,结果表明GM6-1在0.25mol/L LiCl/DMSO溶液中是以无规卷曲的形式存在。Wang等[8]从赤灵芝中分离出5种水溶性的杂多糖(GL-1-GL-V),结果表明,高支化的杂多糖GL-1在0.9%NaCl溶液中为紧凑的线团形式,而随着支化度减小,糖链逐渐变成柔性链。Lai等[39]从赤灵芝中分离得到多糖GLFP和GLMP,当用水作为溶剂时,Huggins常数为0.59~0.93,表明在一定程度上发生了多糖分子的聚集,然而如果用DMSO作为溶剂时Huggins常数则变小,表明多糖分子在DMSO中相互作用减弱,黏度下降。
尽管灵芝多糖的构象特征研究有了一些成果,但关于其分子形态和分子尺寸的数据还较为欠缺,有待于进一步分析和阐明。
4 多糖结构与构象方法学研究进展
4.1 灵芝多糖一级结构分析方案
灵芝多糖中存在多种单糖种类,而连接方式又多样,因此,灵芝多糖的结构分析与小分子或其它生物大分子不同,需要有特殊的方法和技术。通常,多糖一级结构的确定需要得到如下信息:单糖组成、糖苷键的连接方式、糖环的类型、异头构型以及糖残基的连接顺序等,多糖上游离的羟基还会发生取代,如甲基取代、乙酰基取代、硫酸基取代等,这时,也需要提供这些特征基团的信息[40]。因此,多糖化学结构的解析需要结合不同的化学和仪器分析方法进行综合鉴定。综合多糖一级结构的解决方案,图3概括了灵芝多糖结构表征的常用方法及其对应所能得到的信息。
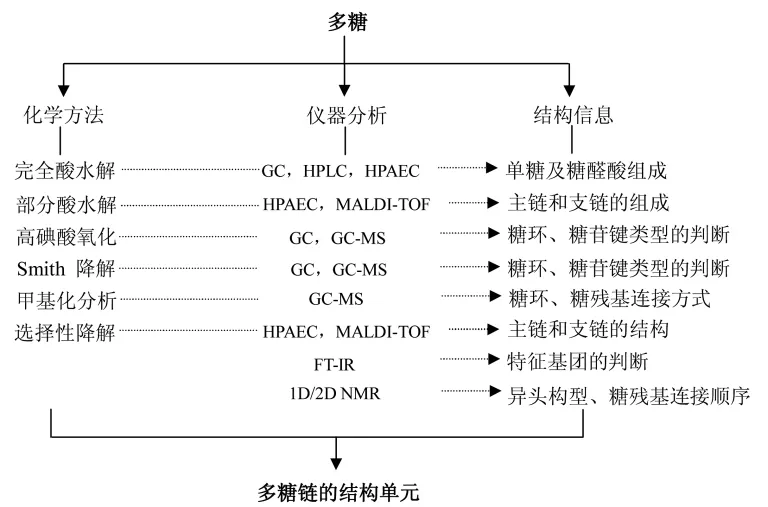
图3 灵芝多糖一级结构分析方案Fig.3 Procedure for the primary structure analysis of Ganoderma polysaccharide
4.2 单糖组成分析
单糖是多糖的最小组成单元,灵芝多糖中常见的单糖包括葡萄糖、半乳糖、甘露糖、岩藻糖、阿拉伯糖、木糖、葡萄糖醛酸和半乳糖醛酸等。要获得单糖组成信息,就需要对多糖样品进行完全酸水解处理,通过强酸溶液(三氟乙酸、硫酸等)在高温下破坏糖苷键,使单糖逐渐游离水解出来,再通过化学方法和仪器分析对单糖进行定性和定量分析。如图3所示,单糖组成分析的方法包括气相色谱法(GC)、液相色谱法(HPLC)和HPAEC-PAD法等。
GC法需要将水解的单糖进行乙酰化,使之能在色谱系统中气化。然而,由于糖醛酸无法进行乙酰化反应,因此,无法直接用GC法进行检测,这时,需要借助强还原试剂(硼氢化钠等)将糖醛酸还原成中性糖,然后进行乙酰化衍生。Wang等[41]通过对多糖的完全酸水解、乙酰化衍生和GC设置进行条件优化,改进了GC分析中性糖和糖醛酸组成的方法。Chen等[42]则建立了基于毛细管气相色谱的黑灵芝多糖单糖组成分析的方法,该法与HPLC法比较,具有更好的重现性和更低的检出限。
HPLC法检测单糖组成同样需要进行柱前衍生,使单糖分子带上紫外吸收基团,从而使之可通过紫外检测器进行检测,常用的衍生试剂是1-苯基-3-甲基-5-吡唑啉酮(PMP)。因糖醛酸可直接被PMP衍生,因此,采用HPLC法测定单糖组成时,不需要进行糖醛酸还原的操作,从而实现中性糖和糖醛酸的同步检测[43]。HPLC法分析单糖组成的操作相对简单,具有更好的灵活性,结合质谱检测器,能有效降低样品的最低检出限。
HPAEC结合脉冲安培检测器(HPAEC-PAD)是单糖组成分析中最常用的方法之一[44],其最大优点是多糖样品经酸水解后,不需要进行衍生化处理,可直接进样分析,从而减小试验误差。通过调节洗脱液的梯度,HPAEC可分离不同种类的中性糖、糖醛酸和寡聚糖等,因此,在多糖的单糖组成分析中应用更为广泛。Zhao等[45]采用HPAECPAD法同步分析了13种单糖和寡糖的混合物;Zhang等[30]则证明HPAEC-PAD法用于分析黑灵芝多糖中的糖醛酸种类时,其结果比GC法更为可信,能够直接证明黑灵芝多糖中的糖醛酸为葡萄糖醛酸。
4.3 多糖的甲基化分析
甲基化分析是灵芝多糖结构分析中常用的一种技术,可用于判断多糖中糖残基的连接方式,其反应原理如图4所示。

图4 β-(1→3,1→6)-葡聚糖的甲基化反应过程Fig.4 Methylation procedure of β-(1→3,1→6)-glucan
总之,多糖分子中游离的羟基与甲基化试剂(常用碘甲烷)发生置换反应,游离羟基上的氢被甲基(-CH3)取代;甲基化完全的多糖分子经酸水解生成部分甲基化的单糖分子,糖苷键连接位点经水解后会形成羟基;通过强还原剂将环状结构的单糖打开之后,对部分甲基化的链状单糖分子进行乙酰化,生成部分甲基化的糖醇乙酸酯衍生物(即PMAA);最后通过GC-MS分析 PMAA,通过 数 据 库(http://www.ccrc.uga.edu/specdb/ms/pmaa/pframe.htm l)比对,确定糖残基的连接方式。
甲基化反应条件非常苛刻,Cui等[40]在Ciucanu和Kerek[46]方法的基础上,在多糖甲基化方面做了大量的工作,建立了一套完善的甲基化方案。概括其有以下几个关键点:
1)样品和试剂需要绝对干燥;
2)用DMSO溶解样品时要使样品彻底溶解,若溶解度有限,可加入适量干燥的氢氧化钠粉末促进溶解,使得游离羟基完全甲基化;
3)甲基化过程尽可能在避光条件下完成,并用氮气保护反应环境。
酸性多糖中含有一定量的糖醛酸,这会在一定程度上影响多糖甲基化的效果,因此,在甲基化前,需要对酸性多糖进行还原。糖醛酸还原常用的还原剂是硼氢化钠,Taylor和Conrad[47]探讨了硼氢化钠还原糖醛酸的动力学过程。而Cui等[40]则认为采用氘代试剂(如硼氘化钠)作为还原剂,能更方便的用于识别被还原的糖残基,如Zhang等[30]采用硼氘化钠还原黑灵芝酸性多糖PSG-1-F0.2,证明PSG-1-F0.2中的糖醛酸连接方式为1→4-葡萄糖醛酸。
不同的PMAA在极性气相色谱柱中能得到有效的分离,因此,可通过GC串联MS检测器来分析不同的PMAA。在相同填充物的色谱柱中,PMAA的重现性非常的好,可通过文献的相对保留时间结合MS信息来推断糖残基的连接方式;然而,在不同填充物的色谱柱上,PMAA的出峰时间不同,因此,在比对保留时间时,需要注意文献数据所用的试验条件。Carpita和Shea[48]总结了不同PMAA在不同色谱柱中的相对保留时间,可作参考。
PMAA在质谱中具有独特的断裂方式,在电喷雾离子源(EI源)下具有一定的断裂规律:
1)PMAA在质谱峰中无分子离子峰,而有一对离子峰的m/z等于这个PMAA的分子质量;
2)碎片离子峰主要来自于PMAA分子中的C-C键断裂;
3)两个带有甲氧基的C-C键最容易发生断裂,其次是一个带甲氧基,一个带乙酰氧基的C-C键,最后是两个都带乙酰氧基的C-C键;
4)碎片离子峰上的正电荷一般在带有甲氧基的碳片断上;
5)碎片离子峰进一步裂分,会按容易程度产生失去 CH3COOH(m/z 60),CH3OH(m/z 32),CH2CO(m/z 42)和HCHO(m/z 30)等二级碎片离子;
6)当C-1被氘代标记时,碎片离子峰中,含有C-1的片断其m/z为偶数,而不含C-1的离子峰m/z为奇数,由此,可以判断异头碳的位置。
甲基化结合GC-MS分析是目前分析多糖糖苷键连接方式最有效的手段之一,在灵芝多糖的结构解析中被广泛使用[18,22,27,29-31,49]。
4.4 多糖的NMR波谱分析
NMR技术在灵芝多糖结构分析中起着非常重要的作用,它可提供异头构型(α或β型)、糖苷键连接方式、糖残基连接顺序等信息,是多糖结构解析中最为关键的一个手段。在1D NMR中,1H和13C NMR最常用[50],多糖在1H NMR中的信号集中于3~6 ppm,很难用于归属糖残基中所有氢的化学位移;而13C NMR的化学位移分布就宽很多(60~110 ppm),具有更好的分辨率。多糖的异头氢出现在4.3~6.0 ppm之间,其中4.3~4.8 ppm为β构型,灵芝多糖的异头氢信号通常出现在这个区域[18,30];而 5.0 ppm 以 上为α 构 型[31];3.2~4.5 ppm之间则为H2-H6信号的集中区域,很难逐一归属;一些特征基团可在1H NMR中判断出来:2.0 ppm附近是乙酰基团(CH3COO-)中氢的特征信号,而1.0 ppm附近则为甲基(-CH3)的氢信号[31]。对于13C NMR,95~105 ppm为异头碳信号,其中β构型异头碳会出现在101 ppm以上,而α构型出现在95~103 ppm之间;65~85 ppm区域为C2-C5的信号集中区,其中,78~85 ppm是C2-C5中取代位置上的碳信号,65~80 ppm区域为未被取代的碳信号;61 ppm附近为未被取代的C6信号,而被取代的C6信号则往低场移至69 ppm附近。另外,对于含有糖醛酸或乙酰基的灵芝多糖,其羰基(C=O)中的碳信号会出现在170~180 ppm的低场,这个特征可用于判断多糖中是否含有羰基化合物[30-31];甲氧基的碳信号则会出现在55~61 ppm之间,乙酰基中甲基上的碳信号会在22 ppm附近,这些特征信号可用于判断多糖中的特殊基团。
多糖中相同原子(尤其是C2-C5和H2-H5)的化学位移差别不大,在1D NMR中会有严重的信号重叠,这时,需要借助2D NMR技术对难以辨认的信号进行归属,由此得到多糖分子的结构信息。2D NMR信息是不同糖残基中C和H化学位移归属和糖连接顺序推断的主要依据。对于多糖,COSY(Correlation spectroscopy),HSQC(Heteronuclear single quantum coherence)和HMBC(Heteronuclear multiple bond coherence)是解析结构过程中必须获得的信息[51]。其中,COSY提供的是同一糖环中相邻氢原子间的偶合关系,即H1-H2,H2-H3,H3-H4,H4-H5和H5-H6之间的偶合,偶合常数越大,峰信号越强。通过糖上容易辨认的氢(如异头氢或甲基氢),结合文献数据,可以顺利的寻找糖上其它位置的氢信号。HSQC可直接获得H和与其相连的C之间的1JCH偶合,在COSY基础上,通过HSQC可以找出糖残基上每个氢所连接的碳信号。HMBC则可获得C和H的远程偶合信号,由此可用于检测相连两糖残基间的C/H偶合,即异头氢(碳)与另一糖残基相连位碳(氢)的偶合,从而推断单糖残基间的连接片断[52]。
随着NMR技术的发展,一些新的2D NMR技术被引入到多糖结构的分析[53]。TOCSY(Total correlation spectroscopy)作为总相关谱,可检测糖分子内异头质子与沿着碳链传递下去的的质子偶合,即H1-H2→H1-H3→H1-H4→H1-H5→H1-H6,随着链的传递,这些信号会越来越弱。TOCSY可以辅助COSY解析属于该偶合链的所有氢信号(主要是重叠比较严重的H2-H6信号)。H,HNOESY(Nuclear overhauser effect spectroscopy)是质子间空间相互接近产生的一种核交叉驰豫现象。在多糖分子中,糖残基连接位点的两个碳上的质子空间位置接近,容易产生NOE信号,因此,对糖残基连接片断的推断有很大帮助[54]。
4.5 多糖的分子质量及分子质量分布
多糖是一种多分散性体系,其分子质量并不仅是一个特定的值,还存在一个分布范围,对于某一特定多糖来说,需要同时给出平均分子质量和分子质量分布这两个信息。在高分子化学中,常用4种统计学方法来描述高聚物的分子质量:数均分子质量(Mn)、重均分子质量(Mw)、z-均分子质量(Mz)和黏均分子质量(Mv)。其中,Mn 可通过膜渗透压法和分子排阻色谱法来测量得到;Mw可通过静态光散射和沉淀平衡法测定,此外,沉淀平衡法还可计算得到Mz;而Mv则需要通过黏度法结合Mark-Houwink方程计算得到[40,55]。对于某一特定多糖,存在以下关系:Mz>Mw>Mv>Mn,其中 Mw/Mn的比值称作多分散性系数,可以用于描述多糖的分子质量分布。对于灵芝多糖,其多分散性系数通常介于1.0~2.0之间,属于宽分布的多分散体系[21,56]。
随着液相色谱技术的发展,分子排阻色谱串联多检测器技术(HPSEC-MALLS)成为多糖分子质量分析的最常用方法之一[57]。通过串联示差(RI)、激光光散射(LLS)和黏度检测器(DP)在线同时检测经分子排阻色谱分离的多糖级分,可获得 Mw、Mw/Mn、Rg和[η]等信息。此外,该方法还可方便的建立Mw与[η],Mw与Rg和Mw与Rh之间的关系,从而获得多糖在溶液中的构象特征[58-59]。Liu等[60]采用HPSEC-MALLS法分析了不同种类灵芝中多糖的分子质量分布,并建立了灵芝中水溶性β-葡聚糖含量测定的HPSEC法,该法对于灵芝多糖的质量控制将起到重要作用。
4.6 多糖的光散射性质分析
光散射是多糖溶液构象分析的重要工具之一,经典光散射技术主要包括静态光散射(SLS)和动态光散射(DLS)[61]。
静态激光光散射是基于Rayleigh散射理论,测定高分子溶液的瞬时光散射强度。物质的光散射强度与样品的浓度(c)和仪器测量角度[P(θ)]有关,通过测定不同角度和不同浓度样品溶液的光散射强度,结合数学拟合方法,可检测高聚物的分子质量、分子链尺寸和样品分子与溶剂之间的相互作用等参数。其中最常用的数学拟合方法是Zimm-plot拟合,此外,针对不同物质的性质,还有Debye、Berry 或 Gunnier等拟合方法[58]。
动态光散射主要依据是物质的布朗运动。分子在溶液中都进行着剧烈的布朗运动,而小分子的布朗运动比大分子的速率要快很多,这在光散射中所表现的就是弛豫时间的差异,小分子弛豫时间短,大分子弛豫时间长。动态光散射通过不同弛豫时间通道,可获得样品在一定时间内的强度时间相关函数(TCF),该函数与样品平移扩散系数(<D>)有关,因此,通过对TCF进行数学拟合,可直接获得样品的<D>,并由此通过Stokes-Einstein方程计算样品的平均流体力学半径Rh[58,62]。
经典光散射由于对溶剂没有限制,在高分子构象分析中具有广泛的应用。结合静态和动态光散射数据,可获得物质在稀溶液中的Mw、Rg、第二维里系数A2、<D>和Rh等构象参数,由此计算高聚物的结构参数ρ(R g/Rh),ρ值是高分子链在溶液中的一个特性参数,能表征高分子链在溶液中的构象特征:在良溶剂中,当ρ=0.77时,为硬球构象;随着ρ的增大,紧凑的高分子链逐渐展开,柔性增强;当ρ=1.5~1.8时,则为线性柔顺链;而ρ接近2时,多分散体系为无规线团或高支化的无规结构;当ρ远远大于2时,多分散体系则为刚性棒状结构[61]。Liu等[13]采用经典光散射分析了灵芝多糖GLP20在不同溶剂中的性质,结果表明GLP20在DMSO中的分子质量是在水溶液中的1/3,结构参数ρ值表明GLP20在水溶液中呈刚性链状结构,而在DMSO中为无规线团,由此推测GLP20在水溶液中存在3股螺旋的刚性结构,而DMSO可以破坏3股螺旋结构,使GLP20分子链展开形成无规线团。
4.7 原子力显微镜
原子力显微镜(AFM)是近年来发展起来的观察生物大分子构象和形貌的有力工具[63-64]。AFM的操作平台有两种成像模式:接触模式和轻敲模式;其工作原理是通过附在一根可活动的微悬臂底端的细小探针来扫描样品,当探针与样品表面有接触时,极微小的作用力即可引起微悬臂的偏转,由此产生相关信号,最后通过光电检测系统对该信号进行检测、放大和转换,得到样品的地形图、相位图、高度图等图像。AFM不同的成像模式对样品具有一定的适用性。接触模式可得到样品的连续结构信号,然而,探针尖端一直与样品表面接触,容易引起样品的漂移或损坏,也容易破坏探针,因此该模式更适用于刚性高分子;而轻敲模式采用高频振动的探针来扫描样品,可控制探针与样品的表面距离,保持振动频率恒定的同时不会破坏样品的表面,因此,轻敲模式对于柔性的生物大分子物质非常重要[65]。
AFM可在接近生理环境的条件下对生物大分子如蛋白质、DNA和多糖等进行形貌扫描。Mcintire等[66]用AFM观察了裂褶菌葡聚糖和硬葡聚糖的三螺旋结构,发现该三螺旋链可通过加热处理解聚形成单股无规线团;Paniagua等[67]则通过AFM技术观察到草莓果胶是一种线型长链的复合物,并存在RGII二聚体,而通过酶解和酸处理,可减小复合物的大小,清除二聚体的形成。由于大部分灵芝多糖的分子都较小,很难通过AFM观察到灵芝多糖的单分子链,然而随着AFM技术的不断发展,其在灵芝多糖链结构的分析中将扮演重要角色。
5 展望
经过近几十年的发展,随着现代仪器分析技术的飞速进步,灵芝多糖结构的研究取得了很大突破。已报道从不同灵芝品种中提取分离得到的灵芝多糖有200多种,其呈现结构多样性、分子质量分布宽等特点。然而,由于灵芝多糖组成复杂,难以分离纯化得到均一的成分,因此对其结构和构象的分析还存在很大挑战。随着分析技术的发展,包括高频NMR(900MHz及以上)、基质辅助激光解吸电离飞行时间质谱(MALDI-TOF MS)、单分子原子力显微镜技术、冷冻电镜、分子模拟等技术在灵芝多糖中应用,将有助于获得更多关于多糖链结构的信息。基于这些技术构建灵芝多糖的结构库,将极大促进灵芝多糖构效关系的构建,并将对灵芝多糖发挥生物活性的内在作用机制和通路的阐明提供理论基础。
