非小细胞肺癌EGFR-TKI耐药机制及应对策略研究进展
2020-01-10姜俊任新玲
姜俊 任新玲
空军军医大学西京医院呼吸与危重症医学科,西安710032
肺癌的治疗正在迅速发展,已从传统的放化疗向驱动突变的靶向疗法发展。表皮生长因子受体(epidermal growth factor receptor,EGFR)的靶向治疗极大改善了患者的预后。亚洲大约50%的非小细胞肺癌(non-small cell lung cancer,NSCLC)患者和西方国家11%~16%的患者存在EGFR驱动突变[1],约90%是外显子19缺失(Exon 19del)或21位外显子L858R点突变(图1)。其他基因组改变如KRAS、BRAF、PIK3CA突变频率较低。EGFR外显子19、20、21和BRAF突变多发生于女性和非吸烟人群,而KRAS突变更倾向于男性和吸烟人群。EGFR、KRAS、BRAF、PIK3CA是可以两两同时存在的[2]。
第一代表皮生长因子受体酪氨酸激酶抑制剂(epidermal growth factor receptor tyrosine kinase inhibitor,EGFR-TKI)如厄洛替尼和吉非替尼,能够可逆性结合EGFR驱动突变Exon 19del、Exon L858及野生型EGFR,其疗效高于EGFR突变NSCLC中的常规化疗[3]。与一代不同的是,二代EGFR-TKI(如阿法替尼)能够不可逆地结合EGFR以及HER2,抑制范围较广谱,但是皮肤胃肠道不良反应加大,而且总体生存率方面并没有显着改善[4]。奥希替尼是美国食品药品监督管理局唯一批准的第三代EGFR-TKI,是ATP竞争性共价EGFR抑制剂。奥希替尼能够高选择性进入中枢神经系统,能够克服一代、二代EGFR-TKI T790M突变耐药问题。奥希替尼与第一代EGFR TKI相比能延长无进展生存期,而且对野生型EGFR的抑制作用小,导致皮肤病和胃肠道毒性的发生率和严重程度降低[5]。奥希替尼尽管具有如此深刻的疗效,但是放在一线治疗还是放在早期EGFR-TKI失败后的后续治疗中还不是很明确[6]。无论是作为一线治疗还是二线治疗,奥希替尼治疗最终都会面临着不可避免的耐药问题。目前就EGFR-TKI耐药机制及其策略概述如下(图2)。
1 一代、二代EGFR-TKI耐药机制
1.1 获得性T790M突变 一代、二代EGFR-TKI最常见耐药机制是EGFR 20号外显子获得性T790M突变。50%~60%的患者应用一代、二代EGFR-TKI后检测到T790M突变。1.1%的患者没有经过EGFR-TKI治疗之前就有T790M原发性突变[7]。原发性T790M突变通常与21L858R共存,而获得性突变通常与19del共存。Oxnard等[8]通过回顾性研究认为,EGFR-TKI耐药可能存在2种T790 M突变,一种以T790M突变为主导的耐药患者,对奥希替尼反应性较好;而另一种以T790 M并存其它耐药机制的患者,对奥希替尼只能暂时获益。
1.2 旁路途径激活 EGFR胞内酪氨酸激酶被EGFR-TKI抑制后,其他酪氨酸激酶受体形成异源二聚体,代偿激活传导下游信号。如HGF/MET扩 增[9]、HER2扩 增、IGF1R/EGFR异二聚体形成[10]、PIK3CA突变、PTEN缺失、KRAS突变[8]、KRAS扩增、AXL/GAS6激活、RET融合突变(KIF5B-RET、CCDC6-RET)[11-12]、BRAF融合突变[13]等。多种信号通路参与EGFR-TKI旁路激活耐药机制发 生,如PI3K-AKT、核 因 子 κB、PLC-PKC、Ras-MEK/ERK等信号通路(图3)。
1.3 小细胞肺癌转化与上皮间质化 在一些EGFR-TKI肿瘤耐药患者中观察到从NSCLC到小细胞肺癌的组织学转化。RB缺失和p53未激活被报道是小细胞肺癌转化机制之一[14],Niederst等[15]在组织类型转化患者中发现RB 100%缺失。
上 皮间质化(epithelial-mesenchymal transition,EMT)是指上皮细胞失去极性和细胞间相互作用,而向间充质表型转变的可逆性过程。大约有1%~2% EGFR-TKI耐药与EMT有关。先前研究表明,AXL激酶、转化生长因子β及IGF1R[16]上调是EMT介导的EGFR TKI获得性耐药的驱动因素。
1.4 其他 厄洛替尼、吉非替尼除了已知T790M突变以及旁路激活耐药机制外,不少新的机制也被发现,如CRKL扩 增、NF-1缺 失、Wnt/β-catenin、m TOR2活 化、激素受体信号通路异常等。
1.4.1 NF-1缺失与CRKL扩增 NF-1是编码神经纤维瘤蛋白的肿瘤抑制基因,是鸟苷三磷酸酶活化蛋白,负性调节p21-RAS信号传导通路。NF-1通过增加鸟苷三磷酸降解速率从而抑制RAS功能。因此,认为其作为肿瘤抑制因子的功能是通过限制正常细胞中的RAS活性而发生的。NF-1缺失激活MAPK通路,并与患者肺腺癌对EGFRTKI的原发和获得性耐药的发生相关[17-18]。体外和体内试验证明丝裂原酶活化蛋白激酶抑制剂降低NF-1的表达能够恢复厄洛替尼的敏感性[17]。
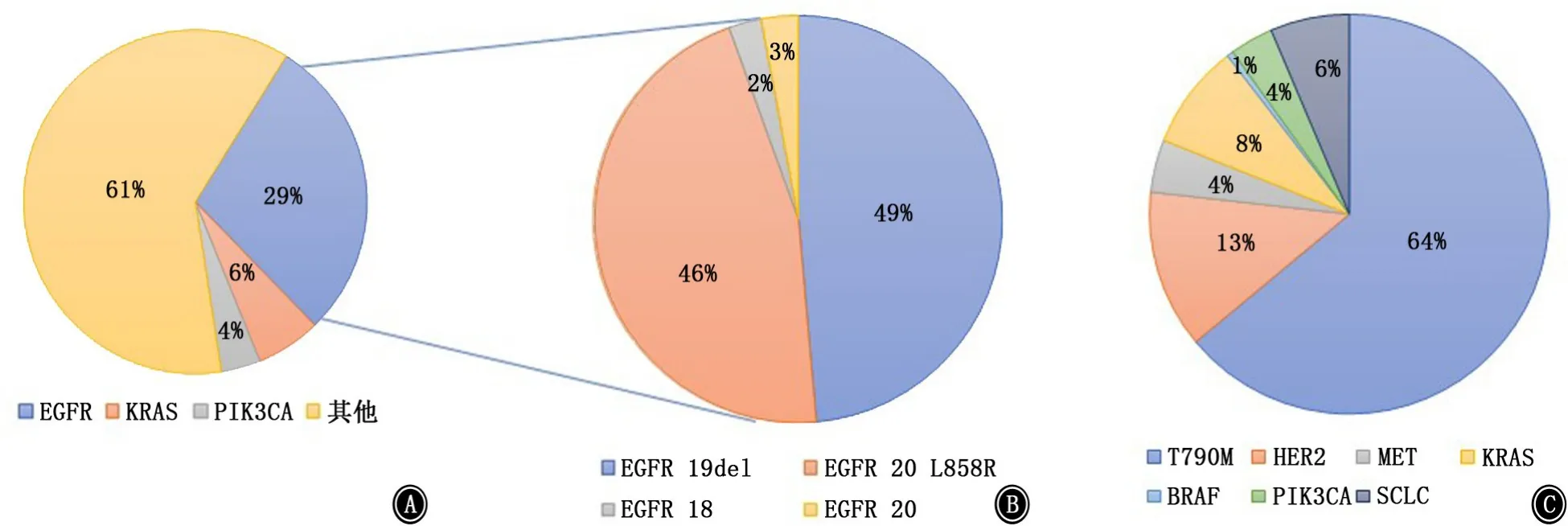
图1 非小细胞肺癌体细胞突变情况及EGFR突变类型 A:非小细胞肺癌驱动突变类型;B:EGFR突变分类;C:一代、二代获得性耐药机制
CRKL是编码衔接蛋白的致癌基因,参与RAS/RAF/MAPK途径的信号转导[19]。在肺腺癌患者中检测到CRKL扩增,并与EGFR-TKI耐药有关。CRKL扩增与NF-1缺失能够协同促进肺癌的发生。CRKL的过表达通过激活SOS1依赖性MAPK和p85依赖性PI3K-AKT信号通路来诱导对EGFR-TKI的耐药性的产生。
1.4.2 β-连环蛋白(β-catenin)激活 Wnt/β-catenin信号通路参与多种肿瘤发生、发展。有报道EGFR-TKI耐药与β-catenin激活有关[20-21]。β-catenin能够维持耐药细胞干细胞特性。肿瘤干细胞对抗癌药物或放射线具有更强的抵抗力并且是肿瘤治疗转移和复发的原因之一,包括肺癌对EGFR-TKI的 耐 药[22-23]。Arasada等[24]报 道 了NSCLC β-catenin诱导耐药的具体机制。β-catenin维持耐药细胞的干细胞特性依赖于Notch3。EGFR在多种肿瘤的发生、发展环节调节β-连环蛋白信号通路。给予EGFR-TKI治疗后,EGFR信号的缺失诱导β-catenin和Notch3的激活。在异种移植模型中β-catenin抑制剂ICG-001和厄洛替尼的组合有更高的抗肿瘤效应,显著延长的肿瘤复发时间和改善总体存活率,并具有良好的耐受性。PRI-724是一种更有效的ICG-001第二代衍生物,目前正处于其他适应症的Ⅱ期试验中,可与厄洛替尼或三代EGFR-TKI联合用于肺癌临床试验。
1.4.3 m TORC2活化 有学者发现m TORC2信号通路对EGFR-TKI耐药细胞的能量代谢重编程现象[25]。肿瘤的葡萄糖代谢和治疗反应密切相关。厄洛替尼敏感和耐药NSCLC细胞系之间代谢有差异,厄洛替尼耐药细胞的生长对葡萄糖剥夺更敏感。耐药细胞具有较低的备用呼吸能力来应对葡萄糖缺乏。在耐药细胞中发现m TORC2磷酸化活化,而m TORC2可通过磷酸化AKT和SGK来调节细胞存活和应激反应。AKT能够整合来自PI3K和m TORC2的信号,并且促进肿瘤细胞生长和增殖。
1.4.4 核蛋白激酶Cδ(protein kinase Cδ,PKCδ)激活PKCδ激活是一代、二代、三代EGFR-TKI共同机制。吉非替尼能够完全抑制EGFR的Y845、Y1068、Y1086位的磷酸化,而只能部分抑制Y1173磷酸化。灭活的EGFR与其他膜受体形成异源二聚化,促进PKCδ核转位从而导致耐药。而灭活EGFR形成异源二聚体能够持续磷酸化Y1173位点,并激活PKCδ。在多种EGFR-TKI耐药肺癌株细胞中发现PKCδ在细胞核内异常激活。而且PKCδ的核转位是耐药的必要条件,抑制PKCδ核转移能够逆转耐药。敲低PKCδ使肿瘤耐药细胞对吉非替尼敏感,而PKCδ的重新表达逆转了这种致敏作用。PKCδ抑制剂sotra和奥希替尼联合用药能够抑制具有抗奥希替尼的T790M阳性肿瘤模型生长[26]。
1.4.5 β2受体激活 肾上腺素能受体通路激活被发现参与了多种恶性肿瘤包括NSCLC的生长及其进展。肾上腺素也能通过激活c AMP来促进肿瘤干细胞的生成,从而导致耐药性的产生[27]。有学者发现肾上腺素和去甲肾上腺素通过激活NSCLC细胞上的β2受体,来抑制肿瘤抑制因子肝激酶B1和上调IL-6表达,诱导EGFR-TKI发生耐药。在EGFR突变肺癌细胞中,β2受体激活下游MAPK信号通路,而MAPK信号通路异常激活是已知EGFR-TKI耐药机制之一[28]。β受体阻滞剂或IL-6抑制剂能减轻抑制肿瘤生长和EGFR-TKI耐药。阿法替尼联用β受体阻滞剂能够给患者带来相对好的收益,PFS能相对延长。
2 奥希替尼耐药机制
不管是原发性还是继发性T790 M突变均对奥希替尼反应良好,但在整个临床治疗期间,获得性T790 M突变的患者在奥希替尼治疗后具有更好的总体存活率[7,29]。同样奥希替尼虽然初始反应率很高,但患者通常也会在治疗后约1~2年后产生获得性耐药。奥希替尼耐药后重新检测T790 M状态是重要步骤,对患者后续治疗策略具有指导意义。约60%患者出现T790M突变丢失伴随其他基因组改变(图2)。
24%~40%的患者给予奥希替尼治疗后会出现三级突变C797S[30]。C797存在于ATP结合口袋中,能与EGFRTKI不可逆地结合。因此,EGFR的外显子20中C797S的点突变影响奥希替尼与EGFR的共价结合位点,从而产生耐药。
EGFR其他少见突变在奥希替尼耐药患者也被发现,如EGFR L718Q[31-32]、G796S、L747P、G724S[33]等。
除了获得性耐药突变,也有原发性耐药机制的存在,如EGFR 19del扩增,以及野生型EGFR扩增[34-35]。奥希替尼其他耐药机制如旁路激活以及表型转变与一代、二代EGFR-TKI相似。
3 EGFR-TKI耐药治疗策略
3.1 EGFR C797S突变耐药治疗策略 获得性C797S突变与T790 M突变以顺式或反式存在。如果C797S与T790M反式存在,第一代和第三代EGFR-TKI联用可能会有抗肿瘤效应;然而,当C797S和T790M突变以顺式存在,患者对任何现有EGFR-TKI都耐药[36-37]。C797S和T790M若从反式向顺式的转变可能是进一步耐药的机制。
3.1.1 JBJ-04-125-02 JBJ-04-125-02是最近研制出的一种可体内外抑制EGFRL858R/T790M/C797S突变阳性肺癌细胞的单一ATP竞争性共价EGFR抑制剂[38]。与单一药剂相比,ATP竞争性共价EGFR抑制剂奥希替尼与突变型EGFR变构抑制剂JBJ-04-125-02的联用导致细胞凋亡增加,更有效地抑制体内外细胞生长[38]。
3.1.2 EAI045 EAI045是第一个针对L858R、T790M及C797S突变设计的变构抑制剂。EAI045对于具有不能形成二聚体的EGFR有更强的活性。联用西妥昔单抗后,二聚体形成被破坏,能完全抑制EGFRT790M/C797S突变阳性肿瘤[39]。
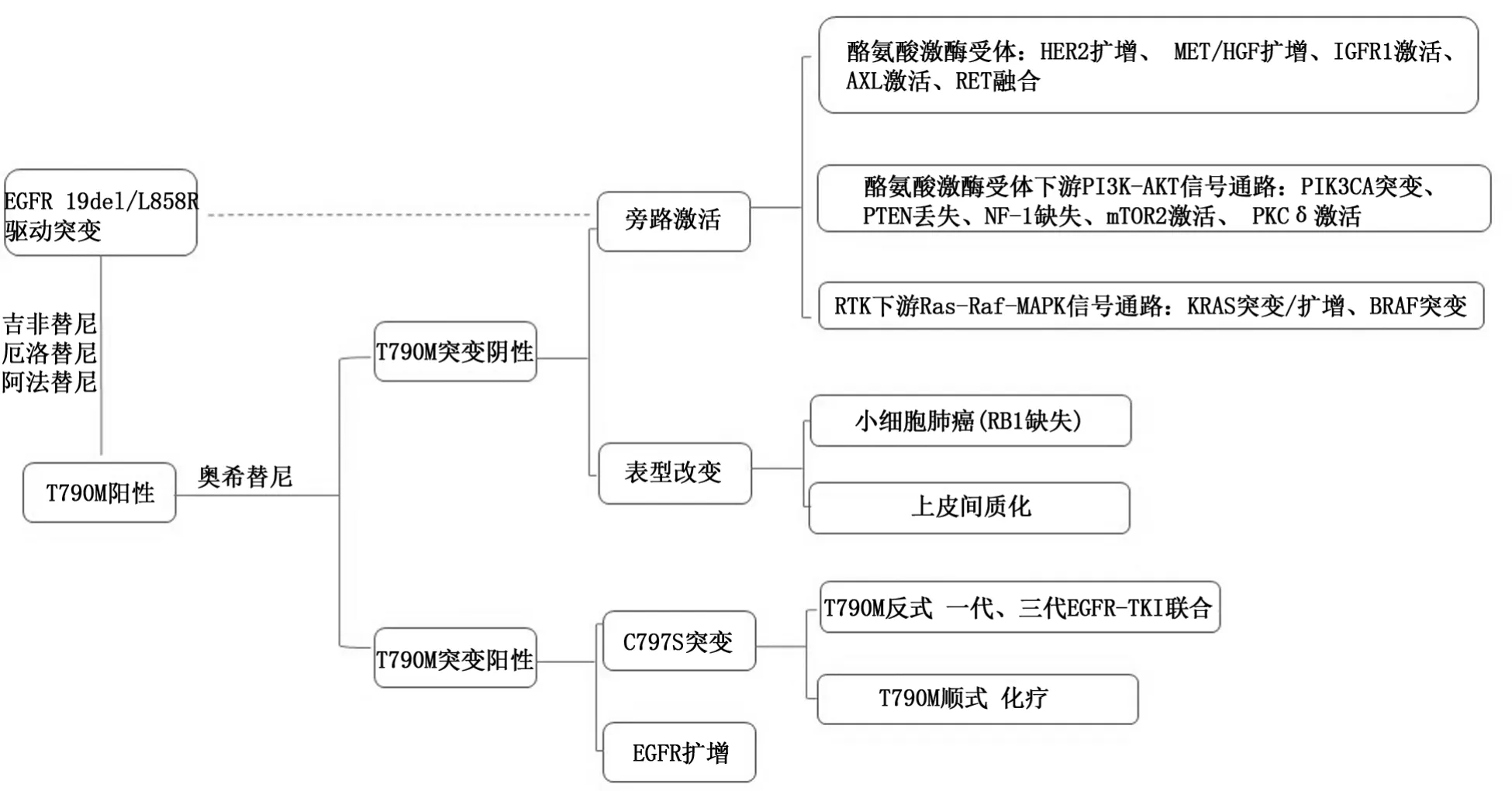
图2 非小细胞肺癌EGFR-TKI耐药机制概要图
3.2 旁路通路激活耐药治疗策略 酪氨酸激酶受体在旁路通路起着关键作用,因此联用小分子抑制剂靶向酪氨酸激酶、单克隆抗体靶向受体或配体,是耐药后的有效治疗策略。长期单独使用EGFR单克隆抗体容易诱发激活其他酪氨酸激酶受体信号通路,如HER2/3的扩增以及刺激HER3与MET相互作用。靶 向HER2、HER3的3联m Ab组合可以阻止下游信号ERK的激活,能够有效抑制肿瘤细胞的增殖与生长[40]。最近有报道3联抗体联用小剂量奥希替尼能够持续抑制消除肿瘤,并可以防止肿瘤复发[41]。
AXL激活是EGFR-TKI获得性耐药机制也是原发性耐药机制[42]。AXL是受体酪氨酸激酶,配体是GAS6。GAS6激活AXL导致MEK/ERK和PI3K/AKT途径激活,使肿瘤细胞对EGFR-TKI的获得耐药[43]。AXL抑制剂NPS1034能够增强耐药细胞对奥希替尼敏感性,并能抑制耐药细胞的产生。在小鼠异种种植肿瘤模型中,NPS1034联用奥希替尼能够抑制肿瘤生长,而单独使用NPS1034对肿瘤增殖没有显著影响。AXL抑制剂的组合用药疗效还需临床实验验证。
根据多臂TATTON试验2个扩增队列的中期结果,MET1/2抑制剂savolitinib联用奥希替尼组合疗法能够给奥希替尼耐药患者带来获益[44]。但是Ⅰ型MET抑制剂savolitinib联用奥希替尼组合同样会产生耐药,在临床1例病例报告中,检测到新的MET激酶结构域突变D1228V;而联用Ⅱ型MET抑制剂cabozantinib能够克服Ⅰ型耐药[45]。selumetinib联用奥希替尼的二线治疗使大多数耐药小鼠恢复了对奥希替尼的敏感性,反应率可达80%。若是组合疗法用于一线治疗,反应率高达90%。selumetinib联用EGFR-TKI也能有效用于KRAS扩增患者。
酪氨酸激酶样孤儿受体1被认为是成年人肿瘤特异性表达的肿瘤胚胎抗原,可以作为肺腺癌治疗的分子靶点。抑制酪氨酸激酶样孤儿受体1的表达可以克服IGF以及HGF旁路信号诱导的EGFR-TKI耐药[46]。敲除酪氨酸激酶样孤儿受体1能够抑制AKT磷酸化,而对ERK没影响,能够抑制由广谱酪氨酸激酶受体引起的肿瘤细胞存活信号通路。
RET为酪氨酸激酶受体,RET融合事件是NSCLC的致癌驱动因子。RXDX-105-01是第一个多中心Ⅰ期试验的RET融合蛋白抑制剂。最新Ⅰ/Ⅰb期试验显示,尽管KIF5B-RET最常见,RXDX-105仅在非KIF5B-RET的 癌症中能够产生应答[47]。临床联用CCDC6-RET抑制剂(BLU-667)和奥希替尼能有效克服耐药。在RET抑制剂vandetanib在治疗CCDC6-RET患者中,有报道发现二次S904F耐药突变[48]。
3.3 表型转变耐药治疗策略 最近有报道EMT转录因子TWIST1通过结合BCL-2的启动子区抑制BIM转录,从而抑制EGFR-TKI引起癌细胞凋亡。而且这一耐药现象可以被BCL-2抑制剂或TWIST1抑制剂盐酸去氢骆驼蓬碱逆转,提示靶向TWIST1或BCL-2可能是克服EMT耐药的有效策略[49]。有学者发现高CD44表达与耐药细胞间充质表型相关,敲低CD44能逆转EMT变化,提示CD44可以预测EGFR-TKI引起EMT的发生[50]。
4 结语与展望
EGFR-TKI耐药的异质性仍然是治疗NSCLC的挑战,治疗过程层出不穷、跌宕起伏。基因组测序技术的进步可以精准检测EGFR-TKI耐药突变情况,有助于后续进一步指导下精准靶向用药。EGFR-TKI获得性耐药问题主要在于EGFR再次突变,并伴有多种酪氨酸激酶受体代偿激活,如MET、HER2、IFGR、AXL等,导致下游AKT、PKC、ERK以及核因子κB等信号通路激活,从而促进肿瘤细胞耐药,进而病情发生进展。联合用药策略是肺癌靶向治疗的未来趋势,能够降低药物副作用同时,也能减少和延缓耐药问题的发生。根据不同的耐药机制来制定适当的个体化治疗策略。因此,进一步基础研究EGFR-TKI耐药分子机制以及耐药治疗策略的临床研究的是至关重要的。
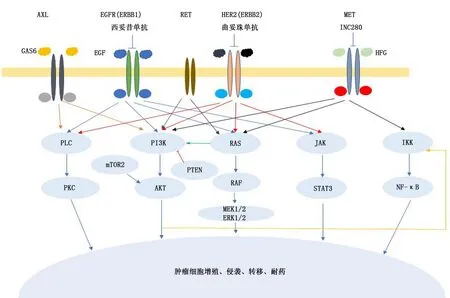
图3 参与非小细胞肺癌表皮生长因子受体酪氨酸激酶抑制剂耐药主要信号通路
利益冲突所有作者均声明不存在利益冲突
