幽门螺旋杆菌诱导的胃腺癌DNA甲基化基因修饰研究进展
2020-01-10谢少洵余钰琨
张 逸 谢少洵 余钰琨 杨 泽 陈 超※
据最新统计,胃癌(GC)在我国癌症的发病率和死亡率排名里均高居第二位[1,2]。胃癌是一种形态异质的癌症,其本身有多种组织学类型,其中胃腺癌占90%,病变部位主要在胃黏膜的腺上皮,是本文的主要研究对象。而胃腺癌在组织学上又分为肠型(IGC)和弥漫型(DGC)两类,前者占胃腺癌的60%,后者占30%[3]。IGC是高分化型,出现于慢性胃炎的环境中[4],它由多种因素引起,其中主要的致病物质是幽门螺旋杆菌(HP)。HP是一种螺旋形革兰氏阴性细菌,尽管人体内只有少数HP会导致癌症发生[5],但实际上大多数的胃腺癌都是由它引起的[4]。HP主要通过诱导表观遗传调控异常来发挥致癌作用[6],本文讨论的DNA甲基化,是一种在发育和疾病中都具有重要作用并且被广泛研究的表观遗传标记[7]。抑癌基因的高甲基化是HP诱导导致的胃黏膜恶变的主要机制,随着疾病的发展,这些基因的异常甲基化会发生在胃腺癌的各个阶段并显示出累积的效应[6,8]。这里所说的阶段是指Correa提出的HP长期感染后发生的多步组织病理学级联反应模式:胃炎,萎缩,肠上皮化生(IM),发育异常,最终癌症发生[9]。对由HP介导的异常甲基化导致的胃腺癌发生过程进行梳理,有助于进一步研究异常甲基化如何影响胃腺癌进程,为胃腺癌的早期检测及治疗提供借鉴。
1. DNA甲基化及异常甲基化概述
DNA甲基化本身是哺乳动物发育所必需的表观遗传学调节过程,这是一种发生在DNA序列中的化学修饰,指的是以S-腺苷甲硫氨酸为甲基供体,在甲基转移酶的作用下,转移甲基到CpG岛的胞嘧啶环的第五个位置上[10]。CpG岛就是富含CpG(胞嘧啶C-磷酸P-鸟嘌呤G)二核苷酸的一些区域,它们主要分布在结构基因的转录起始位点和核心启动子序列中,大约为300~3000bp[6],也有研究表明,位于转录起始位点下游区域,特别是第一内含子,其甲基化程度与基因表达高度负相关[11]。
胃腺癌中的异常DNA甲基化,主要是通过使抑癌基因沉默和激活原癌基因等方式来发挥作用[12],并且这是众多相关基因甲基化的叠加效应。比如对于某些正常细胞的原癌基因来说,他们的启动子中的CPG岛甲基化通常限于稳定沉默的状态,但在癌细胞中会观察到异常的CPG岛甲基化表达[13];而对于在正常细胞中表达的抑癌基因,高甲基化对DNA启动子的异常修饰作用可能导致DNA本身空间结构的改变,影响其稳定性;DNA甲基化也可能影响DNA与蛋白质之间的相互作用,从而致使染色质结构改变,这些都会使得抑癌基因的沉默和转录过程受到抑制,进而会打破体内细胞周期的平衡状态,最后可能诱导细胞异常增殖和肿瘤的发生[8,9]。
2.幽门螺旋杆菌与DNA异常甲基化
有大量研究证明HP与异常甲基化之间有密切的联系。Nakajima等人分析了48个可能在患胃癌时有甲基化迹象的候选基因,其中有26个基因在被HP感染当前或感染过后,其启动子的CpG岛始终保持甲基化[14]。Cheng等人发现:因HP感染诱导的高甲基化发生,使得FOXD3介导的肿瘤抑制因子的转录控制失调,这反过来可能促进胃肿瘤的发展[15];通过医疗手段,彻底根除HP后,可使DNA异常甲基化降低到一定水平并维持一段时间,而目前研究比较多的HP导致甲基化发生机制分为三种。
机制一:通过IV型分泌系统把CagA,肽聚糖和细菌DNA转移到宿主细胞中(图1),这会活化NF-kB,诱导炎症反应发生,进而导致一氧化氮合酶iNOS和白介素IL-1β、IL-8产生,iNOS会催化生成NO,NO又会激活甲基转移酶DNMT,从而诱导抑癌基因的DNA甲基化,最终导致胃腺癌发生[16]。CagA(cytotoxin associated antigen)是一种细胞毒性相关蛋白,它是HP能导致癌症的根源,与CagA阴性HP相比,CagA阳性HP感染在特定基因中诱导更高水平的甲基化。IV型分泌系统是由cagA致病岛编码的,它的作用就是将CagA注射到胃上皮细胞中来发挥其致病性。cagA致病岛全称为细胞毒素相关基因的致病岛,由大约40kbp的DNA片段组成,编码约30个基因包括HP在内的大多革兰氏阴性菌都通过获得被称为致病岛的外源基因簇来发挥其致病性[8,17]。

图1 HP直接作用于胃上皮细胞图
机制二:主要通过HP分泌的CagA诱发炎症反应来发挥作用(图2)。在HP分泌产生CagA和VacA后会诱发胃中的慢性炎症,这会上调白介素IL-6、IL-8、IL-1β与TNF-α、活性氧(ROS)、8-oxodG的表达,在此过程中可能导致相关DNA的高甲基化,从而使得DNA修复基因沉默,其中DNA修复功能被破坏会增加胃上皮细胞的突变率从而促进胃癌的发生发展,VacA(vacuolatingcytotoxin A,空泡毒素)可以抑制细胞增殖并诱导细胞空泡化与凋亡[18]。
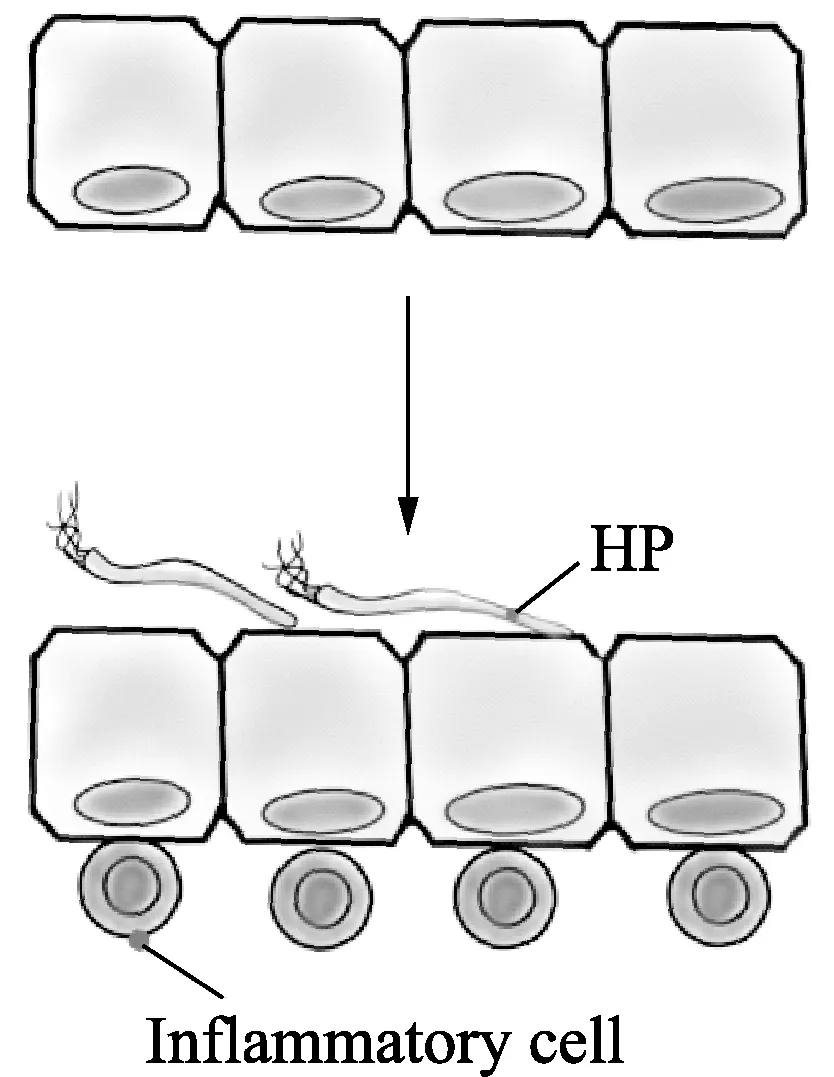
注:在HP将CagA注射到胃上皮细胞后,使得炎性细胞增值浸润,这可能是诱导异常DNA甲基化的更重要因素。图2 HP通过慢性炎症作用图
机制三:从对小鼠的实验中发现了一个并没有炎症反应参与的HP诱导机制。有研究表明,CagA会上调组蛋白-赖氨酸N-甲基转移酶、DNA甲基转移酶3B和c-myc癌基因的表达,从而下调let-7基因的表达,进而诱导胃中原癌基因Ras的表达[16]。
3.异常DNA甲基化与胃腺癌
HP诱导的异常DNA甲基化主要是作用于抑癌基因、致癌基因和DNA修复基因等,并且是众多相关基因甲基化的叠加效应。这些基因分子结构的变化会通过影响DNA转录、细胞周期、细胞凋亡、细胞黏附、原癌抑癌基因表达等生物过程来推动胃腺癌进程(表1)[19]。

表1 Description of common HP-induced genes in gastric carcinoma
3.1 异常DNA甲基化与细胞周期P16基因是调节细胞周期的抑癌基因,位于9p21,其编码的蛋白通过与CDK4和CDK6这两种细胞周期蛋白依赖激酶结合来抑制激酶活性,进而抑制细胞增殖[20]。据最新研究,中国某地区胃癌患者P16基因的启动子区域甲基化率为68.3%[21]。Lee等[22]分别测定了胃癌患者的外周血和肿瘤P16基因的甲基化率,分别为51.9%和66.7%,而正常对照为阴性。以上研究表明P16的启动子高甲基化与HP相关的胃腺癌发生有关。P16的高甲基化导致其表达的沉默,从而扰乱了P16的抑癌作用,和对肿瘤细胞增殖的控制减少,诱导了癌症的恶化。
RASSF1A位于染色体3p21,作为RAS家族的成员,该基因可以通过抑制cyclinD1的积累来阻止细胞周期的进行[20,23]。实际上,RASSF1A主要是通过调节细胞周期和细胞凋亡来抑制肿瘤细胞生长、促进其凋亡衰老的[24]。有研究表明,在胃癌前的各个级联反应阶段都未发现RASSF1A的异常甲基化,而在胃癌中其甲基化率为59%~67%,说明RASSF1A的异常甲基化发生于胃癌晚期[23]。
3.2 异常DNA甲基化与细胞黏附 CDH1是控制细胞间黏性及细胞转移的抑癌基因,位于染色体的16q22.1[23]。其编码的E-钙黏蛋白是一种跨膜糖蛋白,在钙依赖性细胞中起黏附作用[25]。Chan等[26]人从HP感染的消化不良患者的组织样本中发现,CDH1基因的启动子发生了异常甲基化。Huang等[27]人的实验表明HP感染增加胃腺癌细胞中CDH1启动子甲基化,在这个过程中发现HP激活了IL-1β受体和诱导了一氧化氮酶iNOS的表达上调,这催化产生了NO,又发现总DNA甲基转移酶活性增加。CDH1的高度甲基化会使得E-钙黏蛋白表达降低甚至失活,从而导致细胞间黏性降低或出现快速紊乱的现象,最后促进了癌细胞的转移。
VEZT基因位于染色体的12q22,是类似于CDH1的抑癌基因,它编码的VEZT蛋白主要在细胞膜及细胞质中,并且会部分与E-钙黏蛋白连接,起细胞连接控制细胞运动的作用。已有研究表明HP感染诱导正常胃上皮细胞中VEZT的高甲基化沉默[28]。
基因Connexin-32和Connexin-43表达连接蛋白32(Cx32)和连接蛋白43(Cx43),它们是上皮细胞间隙连接的结构组分。有研究发现,这两种基因在癌前期癌性胃黏膜中表达下调,并且在HP感染患者的肿瘤前病变至肿瘤病变的晚期阶段,发现了Cx32和Cx43启动子甲基化增加的现象[29]。
3.3 异常DNA甲基化与细胞的错配修复 hMLH1是调控错配修复的抑癌基因,位于人染色体3p21.3。有研究表明在胃癌中该基因的甲基化率为72.9%,这是导致hMLH1失活的主要原因。hMLH1的表达下降会导致错配修复功能的缺失,这会增加基因组的不稳定性,导致相关原癌基因和抑癌基因的突变概率及微卫星不稳定性增加,最终可能诱导胃腺癌的发生[24]。
3.4 异常DNA甲基化与转录过程RUNX3是位于染色体1q36.1的一种肿瘤抑制基因,也是调节许多癌症相关基因(p27,p53和caspase-3)表达的转录因子,其表达的蛋白RUNX3属于信号通路TGF-β的RUNT区的一个关键点[23]。该基因本身很少有突变发生,而异常甲基化才是导致其失活的主要原因[8,23,24]。有研究发现,在肿瘤前的胃病变中检测到RUNX3启动子的高甲基化,并且显示出从慢性萎缩性胃炎到胃癌高甲基化比例的增加[30]。此外,还发现RUNX3基因因HP引起的胃上皮细胞中启动子高甲基化而失活[31]。
FOXD3基因位于染色体的1p31.3,是Fox转录因子家族的代表,其主要参与自身免疫疾病。FOXD3能激活细胞死亡调节基因RARB和CYFIP2的转录,加之该基因的过表达都会显著抑制胃癌细胞的增殖,促进肿瘤细胞凋亡。因此FOXD3的异常甲基化可能推动癌症的进程。研究显示FOXD3启动子在小鼠和人中的HP相关胃肿瘤中甲基化升高。并且患有高甲基化FOXD3的胃癌患者比其他患者存活时间更短[8]。
3.5 DNA异常甲基化与炎症反应TFF2基因属于三叶因子家族,其位于染色体的21q22.3。TFF2具有调节胃炎症及诱导细胞凋亡的能力,其能上调促凋亡蛋白的表达,下调NF-κB和抗凋亡蛋白Bcl-xL和Mcl-1的表达[8,32]。研究表明,TFF2启动子的甲基化程度在胃肿瘤进展期间增加,并且慢性HP感染与胃黏膜中TFF2基因的启动子甲基化增加相关[8]。
COX-2基因位于染色体1q25.2—q25.3,其编码的环氧合酶-2是一种促炎酶,其在调节胃上皮细胞内稳态和伤口愈合中是至关重要的[8]。研究表明,COX-2的甲基化状态倾向于与COX-2的阴性表达相关,在不表达COX-2的胃癌细胞中检测到该基因的高甲基化水平,说明异常甲基化会调控该基因的表达,并在43.1%的胃癌患者中发现了由于启动子甲基化引起的COX-2的转录沉默[33]。因此,甲基化诱导的COX-2基因沉默可能促进肿瘤前的病理进程。
3.6 DNA异常甲基化与抑癌过程 TFPI-2基因位于染色体7q22的抑癌基因,其编码的蛋白质被称作组织因子途径抑制物2(TFPI-2),也叫作胎盘蛋白-5。TFPI-2能抑制丝氨酸蛋白酶的活性,所以在肿瘤细胞分泌丝氨酸蛋白酶去降解细胞外基质时,TFPI-2能抑制该过程的进行,从而抑制肿瘤细胞的侵袭转移[34]。因此TFPI-2的异常甲基化无疑会促进癌症细胞的迁徙和发展。有研究证实,在胃癌患者的组织中普遍存在TFPI-2高甲基化的现象,但在正常组织和癌旁组织未发现异常甲基化的情况。
除TFPI-2基因外,与胃腺癌相关的抑癌基因APC在与HP感染相关的胃炎中高度甲基化,并且在细菌消除后降低了具有高甲基化APC启动子的病例比例;肿瘤抑制基因WWOX编码含有WW-结构域的氧化还原酶,其经常被发现在几种癌症中被下调,HP感染的胃癌细胞系显示WWOX甲基化程度较高,这与HP的DNMT1和DNMT3表达增强有关;与感染CagA阴性HP的胃癌组织相比,感染CagA阳性HP的胃癌组织中PTEN的表达水平显著降低,并且PTEN在CagA阳性胃癌组织中的表达下降与其启动子甲基化水平的增加有关;此外已经在不同类型的癌症中发现了CYLD(一种肿瘤抑制基因)的失调,已有研究证明,HP菌感染的肿瘤样本中有观察到HP感染与高甲基化和降低的CYLD表达之间的关联[8]。
4.结论与展望
本文依据近年来DNA甲基化诱导胃腺癌的研究报道,从HP诱导DNA甲基化角度深入探讨了胃腺癌发展的分子机制。考虑到胃腺癌虽然是由多因素引起且形态异质的癌症,但HP的感染占主要因素。因此,HP与宿主间必然存在分子遗传学水平的相互作用,HP的感染机制与宿主间表观遗传学分子可能存在必然的联系。HP诱导的异常DNA甲基化主要发生在抑癌基因、原癌基因、DNA修复基因等基因上,这些基因分子结构的改变会导致转录翻译过程异常,基因表达受阻会直接影响细胞的各种生物学过程。在胃腺癌的不同时期,不同的基因功能异常都直接或间接影响肿瘤的病理过程,癌症的最终发生可能是这些基因异常甲基化的合力作用的结果。
近年来,科学家们已经通过实验研究发现了一些受HP诱导的异常DNA甲基化基因,其中一些基因的表达水平已经成为HP感染者的非癌黏膜致癌转化早期指标,并且基因本身已经成为靶向疗法的靶点。未来希望有进一步的研究来揭示发生在胃腺癌不同阶段的DNA甲基化分子作用机制并把它们作为预测胃腺癌的新型生物标志物,这有助于判断患者处于的病理阶段及区分侵袭性肿瘤与相对良性和缓慢生长的肿瘤,从而可以为提出最佳治疗方案提供借鉴。
