黄芪多糖提高脑卒中后抑郁模型大鼠学习能力的实验观察
2019-12-25王元元邓卫平李晓强彭吉霞
王元元, 邓卫平, 李晓强, 彭吉霞
(1. 十堰市太和医院(湖北医药学院附属医院检验部);2. 湖北医药学院基础医学院; 十堰442000)
脑卒中后抑郁 (post-stroke depression,PSD)作为一种继发于脑卒中后的心理障碍性疾病, 具有发生率、致残率和病死率高的特点,患者的核心症状表现为疲劳、淡漠、兴趣减退、情绪低落、反应迟钝及睡眠障碍等为主的心境障碍症状[1]。研究[2]表明,脑卒中病灶以丘脑、海马损伤为主,颞叶、额叶及基底节受损次之。对其治疗过程常伴有不同程度的脑缺血再灌注损伤,其中过氧化物酶体增殖物激活受体-γ(PPAR-γ)表达与脑缺血再灌注损伤后应激反应有关,其水平与PSD有一定的相关性[3]。另有研究[4]表明,PSD的发生与脑海马组织核转录因子-κB (NF-κB)信号通路活性有关。NF-κB信号通路在脑缺血神经损伤可塑性方面发挥着重要的作用,抑制NF-κB活性可减轻缺血性脑卒中大鼠抑郁症状[5]。也有研究[6]表明, 抑郁的严重程度与脑区的脑电振荡电位相关, 海马CA1区自发性动作电位 (AP)和群峰电位(PS)电位降低, PSD抑郁症状明显好转。蒲荣等[7]研究表明, 针刺可显著降低慢性应激抑郁模型大鼠前额叶皮层NF-κB。由此可见,NF-κB信号通路的活性及脑电振荡电位与抑郁的严重程度密切相关,抑制NF-κB信号通路的活性、降低AP和PS电位、纠正脑海马CA1区异常电位可能有利于PSD的康复。基于这一设想,本研究以PSD模型大鼠为研究对象,研究黄芪多糖对NF-κB信号通路及海马CA1区AP和PS电位的影响,分析其对PSD 模型大鼠学习记忆能力及抑郁样行为的影响,为开发新药用于临床治疗PSD提供参考。
1 材料与方法
1.1 动物与分组
SPF级雄性Wistar大鼠48只,体质量(180±10) g, 大鼠由十堰市太和医院生命科学研究所提供[SCXK(鄂)2018-001],在中心标准实验室进行相关实验 [SYXK(鄂)2018-001],随机均分为假手术组、抑郁模型组、黄芪多糖低剂量组(200 mg/kg)和高剂量组(400 mg/kg),每组12只。在实验时,遵守3 R原则,在造模和后期干预性治疗期间给予动物人道关怀,使之充分享受动物福利。实验期间自由饮水和进食,自然光照、保持垫料干燥、动物房恒温(22~24 ℃)恒湿(50%~75%)。
1.2 主要药品及设备
ZH-OM型O迷宫(Zero迷宫)(安徽正华生物仪器设备有限公司); Anymaze分析系统(成都泰盟软件有限公司); 黄芪多糖标准品购自北京索莱宝科技有限公司; 麻醉用水合氯醛购自武汉博菜特化工有限公司; PP-83型玻璃电极拉制仪购自日本Narishige 公司; Bio-Rad酶联检测仪购自美国Bio-Rad公司; 振动切片机和膜片钳放大器均购自英国Campden公司; NF-κB和PPAR-γ试剂盒购自上海超研生物科技有限公司(批号: H1107、H1093); 白介素-1β(IL-1β)和白介素-6(IL-6)酶联免疫吸附试验(ELISA)试剂盒均由上海信裕生物科技有限公司提供(批号: H5034、H5037)。
1.3 PSD模型构建方法及分组
实验前大鼠适应性饲养1 周, 术前禁食不禁水。除假手术组12只外, 抑郁模型组、黄芪多糖低剂量组和高剂量组共36只大鼠,用水合氯醛腹腔注射麻醉, 腹部向上仰位固定大鼠, 剃去颈部被毛, 皮肤用碘伏消毒后铺无菌巾, 切开皮肤, 用玻璃分针分离出左侧颈总动脉和颈内动脉,从颈总动脉(CCA)分叉处插入4-0尼龙线,经颈内动脉(ICA)进入大脑中动脉(MCA), 阻断MCA血供2 h,然后将尼龙线拔出再灌注制备大脑中动脉闭塞(MCAO)模型[8]。在建立MCAO模型的基础上,所有大鼠均单笼孤养,并于造模1周后,每日给予一次慢性不可预见性应激(CUMS), 连续刺激3周以构建PSD模型。具体方法: 按禁食禁水24 h、4 ℃冰水游泳5 min、45 ℃烤箱烘烤5 min、夹尾1 min、水平高速振荡30 min、灯光照射使之活动昼夜颠倒等顺序交替进行应激性刺激[9]。
1.4 干预方法
参考人与动物间药物剂量的换算方法,经计算,大鼠每日用药剂量在200~400 mg。实验人员预实验表明,这一剂量对PSD模型大鼠均产生治疗作用,故正式实验时,黄芪多糖低剂量组(200 mg/kg)和高剂量组(400 mg/kg)大鼠用相应剂量的黄芪多糖灌胃(1次/d),在成功建立PSD模型后灌胃给药治疗 4 周。抑郁模型组和假手术组给予相同体积的生理盐水灌胃对照。
1.5 PSD模型大鼠学习能力及行为学记录
采用糖水摄取实验及Zero水迷宫分析系统评价大鼠抑郁样行为及学习记忆能力[10]。
1.6 脑电振荡电位膜片钳记录
大鼠经水合氯醛麻醉后处死, 去头顶骨暴露脑组织,迅速取出后放入4 ℃通有体积分数95%O2+5%CO2的饱和人工脑脊液中, 分离出大鼠海马CA1区脑组织,修整后用振动切片机切成厚约300 μm的脑薄片。取其中2片于饱和人工脑脊液中孵育 1 h, 用膜片钳放大器记录海马CA1区动作电位(AP)、群峰电位(PS)。
1.7 IL-1β、IL-6的ELISA试验
处死大鼠前取尾静脉血, 严格按ELISA检测试剂盒说明书进行操作, 检测出IL-1β、IL-6浓度。
1.8 CA1区海马脑薄片NF-κB和PPAR-γ蛋白表达
NF-κB和PPAR-γ蛋白表达检测采用Western blotting方法[12]。取未做膜片钳的全部大鼠海马CA1区脑组织制成组织匀浆液, 严格按试剂盒说明书进行操作,检测上述标本中NF-κB和PPAR-γ的吸光度(A)值,然后以标准品的浓度为横坐标、A值为纵坐标绘制标准曲线, 使用curve expert 1.3软件计算出二者相应浓度。
1.9 RT-PCR
采用实时定量-PCR技术检测大鼠海马CA1区脑组织 NF-κB 和 PPAR-γ基因表达[13]。
NF-κB mRNA
上游引物: 5'- AGCCAGTGGAACTGCATGGTAC-3',
下游引物: 5'- GCTAGCCCTGCTAGCTGAG-3',
PPAR-γ mRNA
上游引物: 5'- ACCGACTAAGCGTTCATCTTAC-3',
下游引物: 5'- AGCACTGGCCTCATCTGAC-3',
GAPDH引物: 5'- AGCTCCGATCGGTCCGATTCAG-3',
RNA 抽提试剂盒提取脑组织标本总RNA,然后逆转录,以GAPDH为内参PCR扩增,然后对NF-κB和PPAR-γ基因进行半定量分析。PCR反应条件: 95 ℃预变性2 min,然后95 ℃和52 ℃各30 s、72 ℃ 60 s, 扩增54个循环。以NF-κB和PPAR-γ灰度值/GAPDH灰度值表示NF-κB mRNA和PPAR-γ mRNA的相对值(/GAPDH)。
1.10 统计学处理
采用SPSS17.0软件进行统计学处理,定量资料以表示,多组间比较采用单因素方差分析,组间两两比较采用SNK-q检验,2组间比较采用两独立样本t检验,P<0.05为差异有统计学意义。
2 结果
2.1 Zero水迷宫及糖水摄取
与假手术组比, 抑郁模型组大鼠糖水摄取量明显下降、逃避潜伏期延长, 越台次数减少(P<0.05)。与抑郁模型组比,低剂量组和高剂量组大鼠糖水摄取量明显增多、逃避潜伏期缩短,越台次数增多(P<0.05),且高剂量组与低剂量组比较差异有统计学意义(P<0.05)(表 1)。
2.2 大鼠CA1区海马脑电振荡电位变化
与假手术组比,抑郁模型组海马CA1区AP和PS膜电压明显升高(P<0.05)。与抑郁模型组比较,低剂量组和高剂量组 CA1区AP和PS膜电压明显降低(P<0.05)。高剂量组较低剂量组变化更明显(P<0.05)(表 2)。
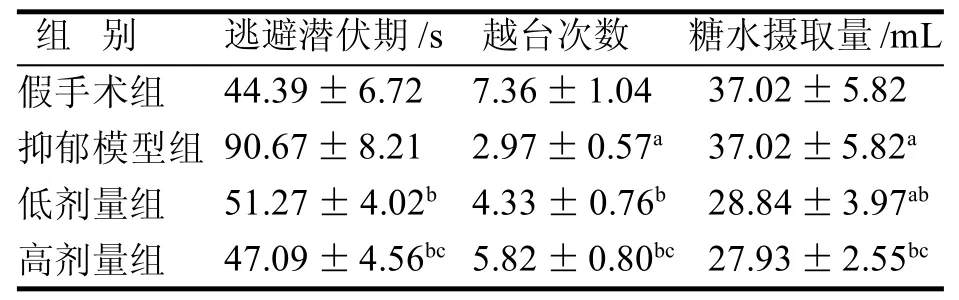
表1 4组大鼠Zero水迷宫及糖水摄取实验结果比较
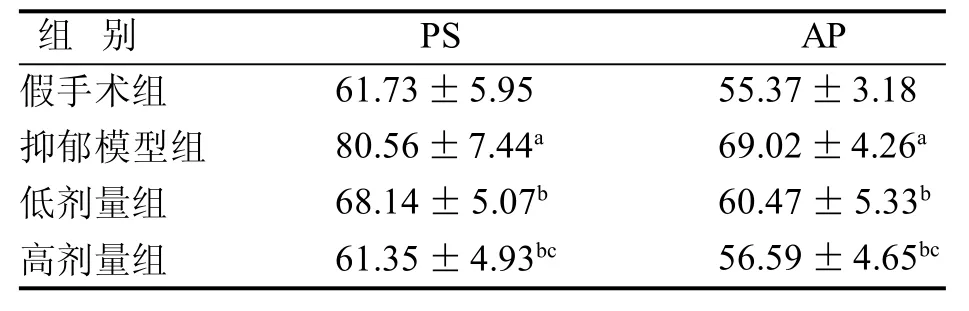
表2 大鼠CA1区海马AP和PS比较 mV
2.3 黄芪多糖对大鼠血清IL-1β、IL-6浓度影响
与假手术组比, 抑郁模型组血清IL-1β和IL-6明显升高(P<0.05); 与抑郁模型组比较,低剂量组和高剂量组血清IL-1β和IL-6明显降低(P<0.05);高剂量组降低更明显(P<0.05)(表3)。

表3 大鼠血清IL-1β、IL-6浓度比较 ng/L
2.4 黄芪多糖对大鼠脑组织NF-κB和PPAR-γ蛋白及其基因表达的影响
与假手术组比,抑郁模型组NF-κB蛋白和NF-κB mRNA表达明显升高,而PPAR-γ蛋白和PPAR-γ mRNA明显降低(P<0.05)。与抑郁模型组和低剂量组比较,Western blotting检测结果显示高剂量组PPAR-γ蛋白及PPAR-γ mRNA高表达、NF-κB 蛋白及NF-κB mRNA 低表达(P<0.05)。高剂量组与低剂量组间比较差异有统计学意义(P<0.05)(表 4)。
3 讨论
PSD是缺血性卒中后最常见的精神病并发症,研究[14]表明,脑卒中2周后27.47%的患者会出现不同程度的抑郁,PSD会延迟卒中后神经功能的恢复,并可能增加卒中复发率和病死率。目前对其病理生理学的研究比较深入,但其发病机制仍然难以捉摸,似乎是多因素的,而不是“纯粹的”心理社会学或生物学改变引起的,大量实验证实, MCAO后创伤性应激可能是造成动物抑郁的主要因素[15]。在PSD病理进程中,存在NF-κB高激活现象,NF-κB信号通路是机体炎症反应的重要体系,是能将信息从胞质传至髓核引起相应基因表达的重要转录因子,主要介导机体组织损伤、细胞分化和凋亡、创伤性应激和防御反应的信息传递,在缺血性脑卒中与血管壁炎症损伤中发挥着重要作用[16]。当机体出现炎症反应时, 处于高激活状态的NF-κB诱导内皮细胞表达一系列趋化因子及黏附分子, 侵入血管内膜形成泡沫细胞, 加速血管形成, 并进一步激活外周血单核细胞, 诱导产生和释放细胞因子IL-1β、IL-6等, 刺激机体产生炎症反应并加重脑损伤[17]。有研究[18]表明, IL-6作为一种重要的炎症介质, 介导PSD病理演变过程中的炎症级联反应,其表达水平与脑损伤程度呈正相关, 而脑损伤程度又与抑郁程度正相关。PPAR-γ具有清除分子氧和氢过氧化物等作用。同时,PPAR-γ可促进NF-κB的磷酸化反应, 从而抑制其活性,因PPAR-γ激活后可以抑制缺血再灌注损伤脑组织及血管中炎性细胞的免疫活性, 且PPAR-γ对巨噬细胞活化起负性调节作用,可抑制单核细胞产生炎性因子IL-1β、IL-2、IL-6等, 从而实现缺血缺氧后对神经元的保护[19]。因此,有理由相信, NF-κB信号通路在PSD的病理演变中具有重要的信号转录作用。调控NF-κB信号通路,提高PPAR-γ活性、抑制NF-κB信号通路的激活可减少IL-1β、IL-6等细胞因子的释放,可降低脑组织炎症反应造成的脑损伤,在一定程度上降低脑卒中后抑郁的发生和发展。

表4 大鼠NF-κB和PPAR-γ蛋白及其基因表达比较
黄芪多糖除具有抗炎、抗肿瘤、增强免疫和降血糖等药理学活性外, 还具有良好的神经营养和神经功能恢复作用, 对改善神经系统相关疾病引起的不同类型的神经损伤具有明显治疗作用[20]。本实验观察发现, 在干预性治疗4周后, 黄芪多糖低剂量组和高剂量组大鼠NF-κB蛋白和NF-κB mRNA表达均明显降低, 而PPAR-γ蛋白和PPAR-γ mRNA明显升高,且血清IL-1β和IL-6明显降低。由此可见,黄芪多糖可能通过提高PPAR-γ活性来抑制NF-κB信号通路活性。NF-κB信号通路活性降低又会引起受其调控的IL-1β、IL-6的释放减少, 这一级联反应对降低脑组织炎症反应造成的脑损伤, 减轻脑卒中后抑郁的发生起到重要的促进作用。这一结果与上述诸多研究结论及王煜等[21]的研究结果高度一致。研究[22]表明,PSD会造成脑组织持续性缺血缺氧,这会导致钠氢通道开放,钠氢通道对维持细胞内pH值以及细胞体积稳定至关重要。钠氢通道过度激活和开放会导致细胞内Na+超载,进而激活钠钙通道,使Ca2+内流增加,这又可能引起细胞内Ca2+超载并造成细胞严重损伤,Ca2+超载是引起PSD患者神经细胞损伤的离子基础。也是抑郁模型组大鼠CA1区海马AP和PS膜电压高于假手术组的离子基础。从膜片钳记录结果看,黄芪多糖低剂量组和高剂量组CA1区AP和PS膜电压较抑郁模型组低,黄芪多糖对离子通道和CA1区膜电位的影响还是首次发现,这一现象提示黄芪多糖具有维持膜电压稳定性的作用。黄芪多糖可能具促进持续开放的钠氢通道关闭,使Na+、H+交换和Na+、Ca2+维持在正常水平,保证细胞内pH值和Ca2+水平的平衡,防止细胞酸中毒并纠正Ca2+超载,促使神经细胞功能得以恢复。黄芪多糖低剂量组和高剂量组大鼠糖水摄取量明显增多、逃避潜伏期缩短,越台次数增多,这一现象提示经黄芪多糖治疗,大鼠抑郁样行为减轻,学习记忆能力增强。这一实验结果提示,黄芪多糖可能是通过抑制NF-κB信号通路的激活而降低受其调控的细胞因子IL-1β和IL-6的释放,减轻炎症反应并提高CA1区膜稳定性,这可能是其治疗PSD的机制之一。
前已述及,PSD引起的抑郁样行为与CA1区海马AP和PS膜电压稳定性有关,本研究的创新点在于在观察黄芪多糖对NF-κB信号通路的影响的同时、结合电生理技术记录其对离子通道和CA1区膜电压的影响并系统分析了可能机制,为其在医药保健等领域的开发和应用方面提供参考,也为研究其它药物治疗PSD提供了可行的研究方法。但PSD是一种“多机制、多因素”的精神病并发症,本研究虽从行为、电生理、生化检测和分子生物学等方面观察黄芪多糖对PSD模型大鼠学习记忆能力及抑郁样行为的影响,但也有不足之处,如未观察MCAO后创伤性应激引起抑郁症状有关的5-羟色胺水平及与机体兴奋性有关的神经递质肾上腺素(Adr)和去甲肾上腺素(NE)水平, 也未观察与神经恢复相关的脑源性神经营养因子(BDNF)和神经生长因子(NGF)蛋白等,并且本实验是在建立PSD模型后给药治疗,而不是在MCAO后和CUMS期间进行预防性给药进行干预性治疗,这不能完全反映黄芪多糖在PSD病理演变过程中的作用,故还不能完全揭示黄芪多糖对PSD的全部可能机制,这也是我们下一步工作的重点。
