以SNAC吸收增强作用为基础的索玛鲁肽片剂研究进展
2019-12-19韩伟杰
李 丹,刘 颜,韩伟杰*
(1.国家药品监督管理局药品审评中心,北京 100022;2.重庆医科大学 药学院,重庆 400016)
据世界糖尿病协会统计,全球共有4.25亿人患有糖尿病,即每11个人中便有1人患有糖尿病[1]。除胰岛素外,胰高血糖素样肽-1(GLP-1)类似物是治疗糖尿病的重要药物。由诺和诺德公司研发的GLP-1类似物索玛鲁肽注射剂型已获得FDA批准上市销售,用于治疗2型糖尿病和肥胖症。然而注射型抗糖尿病药物依从性差的问题一直制约着此类药物使用[2]。因此,研究人员一直致力于开发抗糖尿病药物的口服给药途径。目前,索玛鲁肽片剂的三期临床试验PIONEER已结束,诺和诺德公司宣布将于2019年第一季度末向FDA提交索玛鲁肽口服剂型的上市申请[3]。本文通过检索文献,综述索玛鲁肽片剂的药物代谢动力学及临床试验情况。
1 索玛鲁肽片剂
长效GLP-1受体激动剂(GLP-1RA)主要通过3种机制产生血糖调节功能:以葡萄糖依赖的方式刺激胰岛素释放,在高血糖时抑制胰高血糖素活性,及胃排空的轻微延迟导致葡萄糖吸收减慢[4]。索玛鲁肽为多肽化合物,分子量大,有亲水性,渗透性低[5]。口服递送多肽类药物的主要挑战之一是,这些化合物很难通过胃肠道黏膜被人体吸收。胃中含有较多胃酸及胃蛋白酶,索玛鲁肽在胃中易被降解,失去其药理活性。已有试验观察到,索玛鲁肽单独口服使用在人体中的吸收量难以达到有效的浓度,吸收量在不同个体及同一个体不同服药过程中存在极大的变异性[6]。诺和诺德公司尝试利用合成物N-(8-[2-羟苯甲酰氨基])辛酸钠(SNAC)作为渗透增强剂与索玛鲁肽共同制成片剂,开发出用于口服给药的索玛鲁肽片剂[7]。
1.1 吸收部位
索玛鲁肽片剂中不含崩解剂,其在胃中的分解以表面侵蚀的方式进行[7]。在一项交叉试验(NCT01619345)设计中使用两种水量(50和240 ml)送服含核素的药物。γ相机闪烁扫描动态成像结果显示,在禁食状态下单剂量口服索玛鲁肽(10 mg,含300 mg SNAC;含111 In标记离子交换树脂)后,用240 ml水进行闪烁照相成像,所有受试者均完全出现片剂崩解[8]。为了证明吸收部位在胃,在补充进行的动物试验中,经幽门结扎处理的犬只胃内注射索玛鲁肽,其血浆索玛鲁肽暴露量与使用索玛鲁肽片剂的未结扎犬相当;未结扎犬胃内给药后脾静脉的浓度较高,表现为脾静脉与门静脉药物暴露量(AUC)的比值为1.94(95 % CI 1.15~2.74;P<0.05)[9]。
1.2 SNAC
SNAC为索玛鲁肽片剂的吸收促进剂,是由水杨酸合成的N-乙酰化氨基酸衍生物,有弱酸性和两亲性。SNAC是唯一被批准用于改善口服途径生物利用度的渗透增强剂[10]。在其他临床前研究中,SNAC有改善肠道内多肽(如胰岛素)渗透的作用[11]。索玛鲁肽片剂置于不同体积的模拟胃液中,pH值由酸性逐渐变为中性,体积越小,则缓冲作用越明显;相反,不含SNAC的索玛鲁肽片剂对模拟胃液pH无明显影响[9]。在胃蛋白酶存在的条件下研究不同pH下索玛鲁肽片剂稳定性的变化。模拟胃液pH为7.0时,假定索玛鲁肽降解遵循一级代谢动力学,索玛鲁肽半衰期超过100 min,而当pH为5.0和2.6时,索玛鲁肽半衰期仅为34和16 min[9]。可以推断,SNAC能在胃中发挥缓冲作用,提高局部胃上皮表面的pH。SNAC的缓冲作用使酶活性降低,索玛鲁肽在胃液中降解时间延长,有利于被人体吸收。
1.2.1 索玛鲁肽与SNAC SNAC提高索玛鲁肽在胃上皮模型(NCI-N87)表观渗透率的作用程度随时间变化。在含索玛鲁肽的胃上皮模型中添加SNAC,短时间内索玛鲁肽表观渗透率相对于未添加SNAC时显著增加(10 min,P=0.008),随着时间延长,SNAC对索玛鲁肽表观渗透率的提高作用减弱(30 min,P=0.142;60 min,P=0.568)。以不同时间间隔向含索玛鲁肽的胃上皮模型中添加SNAC,SNAC增强渗透的作用程度也不同。间隔时间为10 min时,索玛鲁肽表观渗透率为2.06±0.53,间隔时间为30 min的索玛鲁肽表观渗透率为1.30±0.27。间隔时间越长,SNAC对索玛鲁肽表观渗透率的增强作用越弱[12]。
SNAC对索玛鲁肽有渗透增强作用且具有浓度依赖的特征。胃上皮模型暴露于20 ~ 60 mmol/L SNAC时,该组索玛鲁肽表观渗透率低于无SNAC的胃上皮模型对照组。而当模型中SNAC浓度达70 mmol/L时,索玛鲁肽表观渗透率相对于无SNAC的胃上皮模型对照组几乎翻倍,浓度达80 mmol/L时甚至达到7倍[9]。
在喂食索玛鲁肽片剂的麻醉犬体内,使用内镜吸取距离片剂中心0,3,6 cm处的液体测量其所含药物浓度。结果表明,30 min后距片剂中心6 cm处胃上皮表面药物浓度较片剂下方(0 cm)浓度显著减小(索玛鲁肽,P=0.007;SNAC,P=0.008)。随着与片剂距离增加,索玛鲁肽与SNAC浓度显著减小。胃镜观察到,胃组织对索玛鲁肽免疫反应仅发生在片剂下方及其周围的上皮表面。胃减少片剂的稀释与扩散,使其在胃表面局部集中释放。因此,SNAC与索玛鲁肽的共剂型,而不仅仅是共给药,是实现高效吸收增强的必要条件[9]。
1.2.2 机制研究 索玛鲁肽分子有两亲性,分子间可通过疏水相互作用互相结合形成低聚物。索玛鲁肽片剂在溶解后,SNAC可引起溶液极性变化导致分子间疏水相互作用减弱。应用核磁共振波谱法(NMR)、动态光散射(DLS)等技术观察到,索玛鲁肽在SNAC的影响下表观分子量减小,分子由低聚物状态朝着单体化方向转变[9]。
SNAC有亲脂性,能有效地插入胃上皮质膜,从而改变胆固醇、磷脂和蛋白质固有的包装完整性,进而影响膜的流动性[13]。应用高灵敏度差示扫描量热法观察到,随着SNAC浓度增加,二肉豆蔻酰磷脂酰胆碱(DMPC)膜转变热逐渐降低[9]。使用SNAC的正异构体o-SNAC作为渗透增强剂,服药后180 min内索玛鲁肽血浆暴露量(AUC0-180min)与使用SNAC作为渗透增强剂相比差异大(P< 0.001)[9]。异构体o-SNAC的吸收增强作用明显减弱,可能与o-SNAC中羟基的位置改变,苯环中脂肪酸的方向和电子分布也发生变化,导致其插入细胞膜的倾向发生改变[9]。
乙二胺四乙酸(EDTA)可通过螯合作用消耗细胞外钙激活PKC通路,导致细胞外途径的扩张[14]。在胃上皮模型(NCI-N87)分别添加EDTA与SNAC观察其对索玛鲁肽渗透过程的增强作用,同时设置一不添加任何渗透增强剂的胃上皮模型作为对照组。与对照组表观渗透率相比,EDTA与SNAC(SNAC浓度为80 mmol/L,EDTA浓度为75 mmol/L)对渗透过程的增强作用显著(P<0.001)且程度相似。检测索玛鲁肽在细胞内的积累量,EDTA与对照组相比无显著性差异(P=0.057),而SNAC较对照组差异显著(P<0.001)[12]。可以推测,SNAC增强索玛鲁肽渗透作用主要通过跨细胞途径而非细胞外途径。电子显微镜观察索玛鲁肽口服给药后小鼠胃黏膜上皮支持这一判断[9]。
2 药物代谢动力学
索玛鲁肽片剂的药物代谢动力学参数见表1[15]。Granhall等[16]发现,健康男性和2型糖尿病男性患者血浆中索玛鲁肽暴露量(AUC)和最大浓度(Cmax)相似,其估算比率分别为1.00(95 % CI,0.60;1.66)和1.00(95 % CI,0.61;1.62);在健康男性和2型糖尿病男性患者中,10周内索玛鲁肽片剂一日一次剂量达40 mg是安全且可耐受的。
肝肾功能损伤不影响索玛鲁肽药物代谢动力学过程。索玛鲁肽经肾排泄很少。在一项研究肾功的临床试验中,仅有一例患有终末期肾病的受试者尿液中检测到索玛鲁肽[17];而在一项研究肝功能的临床试验中,所有受试者尿液样本均未检测出索玛鲁肽[15]。

表1 索玛鲁肽片剂在正常人体内的药代动力学参数
研究发现,速尿和瑞舒伐他汀与索玛鲁肽片剂合用时血浆暴露量增加,而SNAC对其无影响,研究人员判断这一现象可能与GLP-1成分已知的胃排空延迟效应有关[18]。此外,索玛鲁肽片剂对二甲双胍和地高辛的血浆暴露量无临床相关影响[19],也不影响口服避孕药炔雌醇和左炔诺孕酮的生物利用度[20]。
2.1 肾损害
在一项多中心、开放标签、多剂量、对照试验(NCT02014259)中,受试者依据Cockcroft-Gault公式计算所得的肌酐清除率分组,有正常组(n=24)、轻度损伤组(n=12)、中度损伤组(n=12)、重度损伤组(n=12)与终末期肾病组(n=11,需在服药后接受血液透析治疗)。各肾功能损伤组药动学参数相对肾功能正常组的估算比率(90 % CI)如表 2所示[17]。
由表 2可见,肾功能损伤对索玛鲁肽片剂在血浆中的暴露量无一致的作用模式;同时也支持,个体对索玛鲁肽片剂的吸收程度存在差异[6]。除终末期肾病组外,SANC的吸收程度随着肾功能损伤程度的增加而增大。
在接受血液透析治疗后,终末期肾病受试者血浆索玛鲁肽暴露量与根据透析规律所预测的结果相似,两者比值接近于1(95 % CI 0.92;1.01);而透析与非透析情况下SNAC血浆暴露量的比值为0.95(95 % CI 0.74;1.23)[17]。由此可见,血液透析对索玛鲁肽与SNAC的药动学过程无影响。
2.2 肝损害

表2 各肾功能损伤组药动学参数相对正常组的估算比率(90 % CI)
在一项多中心、开放标签、多剂量、对照试验(NCT02016911)中,研究者将受试者依据Child-Pugh标准划分为肝功能损伤轻度组(n=12)、中度组(n=12)、重度组(n=12)与肝功能正常组(n=24)。试验中采集血样获取片剂在体内的药动学参数并计算各组间的估算比率(见表3)。结果表明,各肝功能组索玛鲁肽吸收情况相似,无显著差异。随着肝损伤程度的增加,个体对SNAC吸收增加[15]。
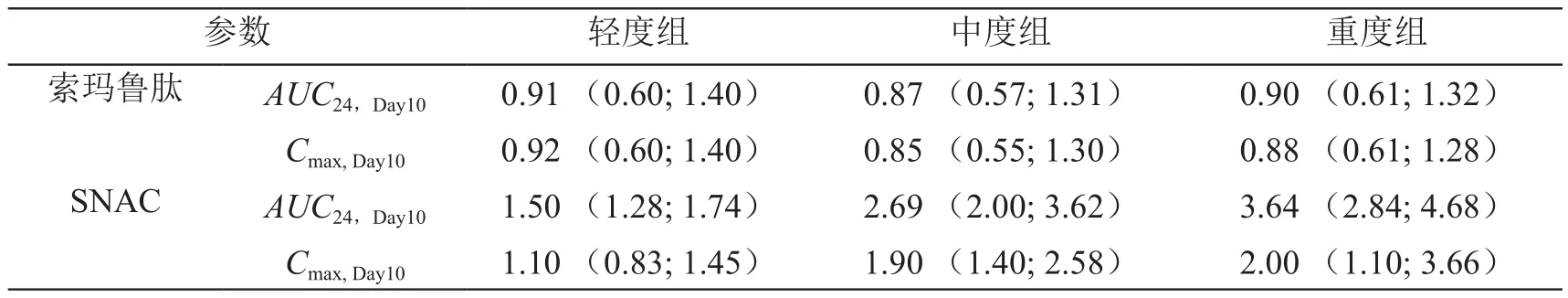
表3 各肝功能损伤组药动学参数相对正常组的估算比率(90 % CI)
3 索玛鲁肽片剂的用法
索玛鲁肽片剂在胃中以表面侵蚀的方式分解并被人体吸收[7],也更易受胃生理学因素、内容物等的影响。研究表明,GLP-1类似物有延迟胃排空速率的作用,这一特性也使得索玛鲁肽片剂在胃内滞留时间延长,血浆暴露量增加[21]。SNAC中和胃酸可促进索玛鲁肽吸收,影响胃酸也可对索玛鲁肽片剂吸收产生影响[9]。诺和诺德公司也资助了许多针对索玛鲁肽片剂用法的临床研究。
3.1 食物
索玛鲁肽片剂应在禁食状态下服用,以减少胃中食物阻碍。一项评估食物对索玛鲁肽片剂影响的临床试验(NCT02172313)中,将受试者分为进食组(240 ml水送服,服药前30 min摄入的高热量、高脂肪早餐,服药后4 h内禁食)、禁食组(240 ml水送服,服药前通宵禁食至少10 h,服药后4 h内禁食)与对照组(120 ml水送服,服药前通宵禁食至少6 h,服药后30 min内禁食)3个组别。禁食组所有受试者均能检测到可测量的索玛鲁肽血浆浓度,而进食组25名受试者中有14名受试者未观察到可测量的索玛鲁肽血浆浓度,其余11名受试者只观察到有限的药物血浆浓度。禁食组索玛鲁肽血浆暴露量(95 %CI;1.41 [0.96;2.07],P=0.082)和最大血浆浓度(95 %CI;1.41 [0.96;2.08],P=0.080)高于对照组,但无统计意义上显著性。禁食组达到最大血药浓度的时间相比对照组延长(中位数分别为1.75,1.00 h),但半衰期无明显差异(几何平均数分别为160 ,152 h)。120 ml水和30 min的服药后禁食时间有望被患者接受[22]。
3.2 水
受试者在禁食状态下分别用50或140 ml水送服含111In的索玛鲁肽片剂并禁食4 h,γ相机闪烁扫描动态成像结果表明,50 ml水送服片剂在受试者体内发生完全分散的平均时间为85 min,而140 ml水送服时的平均时间为57 min。不同体积水送服片剂致其完全崩解的时间有所不同,但无明显的统计学差异(95 %CI 1.51 [0.96;2.37])。研究表明,片剂侵蚀速率越慢,索玛鲁肽血浆暴露量越高[21]。在另一项试验(NCT01572753)中也发现,给药时两种不同体积(50,120 ml)的水对索玛鲁肽血浆暴露量(AUC)无显著差异(P=0.54),对血浆最大血药浓度(Cmax)也无显著差异(P=0.68)[23]。
3.3 空腹时间
一项试验(NCT01572753)在受试者服药后设置了不同的空腹时间(15,3,60,120 min)。给药方案结束后检测受试者血浆索玛鲁肽暴露量(AUC0-24h,Day10)与最大浓度(Cmax,Day10)。经统计分析,随服药后空腹时间延长,如空腹时间在15与30 min时,血浆中索玛鲁肽暴露量与最大血药浓度显著增高(P值均小于0.001);索玛鲁肽达峰时间也随空腹时间延长而增加。但空腹时间与索玛鲁肽半衰期无明显临床相关性[23]。
3.4 服药间隔与不良反应
研究人员发现,胃肠道副反应如恶心等发生,与索玛鲁肽片剂的使用有因果关系[17]。为减轻胃肠道不良反应,临床试验设计的给药方案通常为10 d剂量递增方案。这一方案也减少了单剂量给药出现的个体药物吸收差异[6]。在索玛鲁肽片剂的II期临床试验中,当患者开始服用较低剂量索玛鲁肽片剂时,恶心事件较少;随着治疗的继续,大多数患者的恶心发生率和严重程度下降,部分原因是一些患者因为这些事件而过早停止治疗。试验中发现,与每4周递增剂量方案组(23 %)和每2周递增剂量方案组(26 %)相比[24],每8周递增剂量的方案组由于不良反应导致的过早停药患者比例更低(14 %)。
4 有效性
4.1 临床试验
一项为期26周的II期临床试验(NCT01923181)研究了索玛鲁肽片剂的临床有效性。为减少单次给药后索玛鲁肽血浆暴露量的变异性,避免药物浓度均低于定量下限(LLOQ),试验设计了连续10 d给药方案,给药剂量每8周(剂量缓慢升高组)、4周(剂量标准升高组)或2周(剂量快速升高组)加倍剂量,直至达到维持剂量。索玛鲁肽片剂2.5 mg标准升高组平均糖化血红蛋白水平下降0.4 %,5 ~ 40 mg标准升高组下降1.2 %~1.9 %,而安慰剂组下降0.3 %,差异显著(95 %CI;2.5 mg组P<0.01,5 ~ 40 mg组P<0.001);而其受试者平均糖化血红蛋白≤7.0 %的比例也显著高于安慰剂组(95 % CI;2.5 mg组P=0.04,5~40 mg组P<0.001)。结果还表明,40 mg 索玛鲁肽片剂标准升高组与皮下注射索玛鲁肽1 mg组在糖化血红蛋白控制方面无显著差异(P=0.80)。索玛鲁肽片剂剂量越大,药理作用越显著。空腹血糖水平下降主要发生在4~8 周。索玛鲁肽片剂10~40 mg标准升高组体重下降4.8~6.9 kg,皮下注射索玛鲁肽1 mg组体重下降4.2 kg,与安慰剂组相比差异显著(95 % CI;P<0.001)[24]。
PIONEER是索玛鲁肽片剂的Ⅲa期临床研究项目,期间共有8845名2型糖尿病患者参与了10项临床试验,全部于2018年完成[3]。PIONEER项目主要指标为糖化血红蛋白,其研究目的与结果汇总如表4、表5所示[25-33]。
4.2 心血管
PIONEER6(NCT02692716)是一项多国、随机、安慰剂对照、双盲试验,针对有高心血管疾病风险的2型糖尿病患者评估索玛鲁肽片剂的心血管安全性。受试者随机分为两组接受索玛鲁肽片剂与安慰剂治疗。为减轻胃肠道不良反应,患者服用剂量逐渐增加,最终达14 mg并维持至试验结束(每隔4周增加一次剂量,分别为3,7和14 mg)。患者被要求在早晨处于禁食状态下服用片剂,服用片剂时水量约120 ml,服药后禁食或停止服用药物至少30 min。主要终点是从随机化到首次发生主要心血管不良事件的时间,包括心血管死亡、非致命性心肌梗死或非致命性卒中。3 183名有心血管风险的2型糖尿病患者登记参加试验。84.6 %受试者年龄大于50岁且有心血管疾病与中度脑血管疾病既往史,15.4 %受试者年龄大于60岁且存在至少一个心血管疾病发作相关风险因素[34]。
索玛鲁肽注射剂型的心血管安全性已经得到证实[35],因此PIONEER6计划的试验时间较少,随访时间中位数为16月。试验中共积累了137例主要心血管不良事件。与安慰剂组相比,索玛鲁肽片剂的相对危险度为0.79,未达到统计学意义上的显著区别。索玛鲁肽片剂显著降低了心血管死亡发生率(相对危险度 0.49,P=0.03),但非致命性心肌梗死与非致命性卒中的发生率降低不显著(相对危险度分别为1.18与0.74)。此外,索玛鲁肽片剂显著降低了49 %的全因死亡率(相对危险度 0.51,P=0.008)[36]。

表4 索玛鲁肽片剂PIONEER临床试验设计方案

注:*前26周索玛鲁肽3,7,14 mg与安慰剂组共同使用的胰岛素剂量分别为60,62,54,56 U/d,后26周剂量调整量分别为+2,-6,-7,+10 U/d;**9 %受试者剂量为3 mg,31 %受试者剂量为7 mg,60 %受试者剂量为14 mg
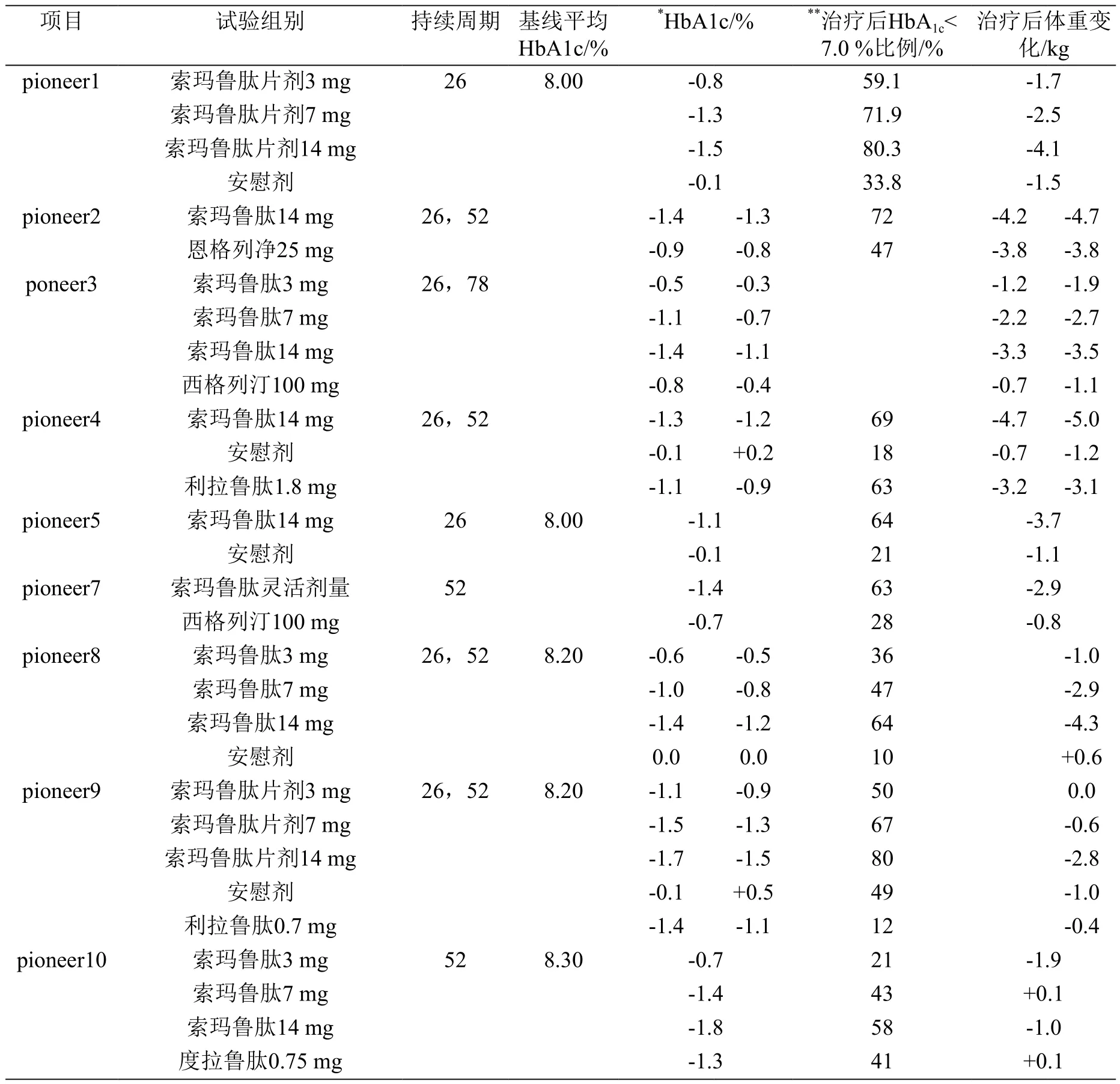
表5 索玛鲁肽片剂PIONEER临床试验结果汇总
4.3 日本人群
PIONEER9是一项随机、双盲、安慰剂对照、开放标签、主动控制的3期临床试验,目标受试者为日本成年2型糖尿病患者。本次试验招募243名受试者接受单药治疗。受试者随机分为5组分别服用索玛鲁肽3,7,14 mg,安慰剂及利拉鲁肽0.9 mg,同时,受试者有接受饮食和运动治疗,或口服抗糖尿病药物作为单一治疗的补充。52周时,安慰剂组平均血红蛋白升高0.5 %,利拉鲁肽组降低1.1 %,索玛鲁肽7,14 mg组分别降低1.3 %,1.5 %。索玛鲁肽3,7,14 mg组平均血红蛋白<7.0 %比例分别为50 %,67 %,80 %,而利拉鲁肽组与安慰剂组分别为49 %,12 %。索玛鲁肽14 mg组在52周后体重显著下降2.8 kg,而安慰剂组为1.0 kg, 利拉鲁肽组为0.4 kg。试验中,索玛鲁肽片剂耐受性良好,安全性符合GLP-1类似物预期。索玛鲁肽组最常见的不良反应是便秘和轻度至中度恶心,且随着时间的推移,这些不良反应逐渐减轻[32]。
PIONEER10研究了索玛鲁肽片剂在至少一种口服降糖药不能充分控制血糖的日本2型糖尿病患者中的有效性。试验还设置了度拉鲁肽0.75 mg组进行对比。从基线8.3 %开始,索玛鲁肽片剂14 mg组受试者在52周后平均糖化血红蛋白显著降低1.8 %,而度拉鲁肽0.75 mg组受试者平均糖化血红蛋白显著降低1.3 %。口服3和7 mg 索马鲁肽的患者糖化血红蛋白分别下降0.7 %和1.4 %。21%,43 %和58 %的受试者在口服索玛鲁肽片剂3,7和14 mg治疗后糖化血红蛋白低于6.5 %,而在度拉鲁肽0.75 mg组中,这一比例为41 %。与基线相比,52周索玛鲁肽片剂14 mg组受试者的体重减轻1.9 kg,有统计学上显著性[33]。
5 安全性
在PIONEER III期临床试验中,索玛鲁肽安全性和耐受性良好,与GLP-1为基础的治疗相一致。无致命的事件的发生。几乎所有报告的不良反应均为中度或轻度,随着剂量的增加,不良反应的严重程度呈上升趋势。由于不良反应,索玛鲁肽片剂(6 %~27 %)相比安慰剂(1 %)发生过早停药的比例也更高[24]。不良反应发生率或严重程度与肝功能损伤程度无明显规律[15]。
5.1 胃肠道疾病
在已完成的临床试验中,最常见的不良事件为轻至中度胃肠道疾病(包括恶心、呕吐、腹部不适等)。研究人员认为其与索玛鲁肽片剂的使用有因果关系[17]。在II期临床试验中,各索玛鲁肽片剂组(31 %~77 %)和索玛鲁肽注射剂组(54 %)报告胃肠道疾病的受试者比例高于安慰剂组(28 %)[24]。在PIONEER Ⅲ期临床试验中,最常见的不良事件都是轻度到中度恶心,随着时间的推移,恶心症状逐渐减轻[25-33]。索玛鲁肽片剂PIONEER项目不良反应汇总见表6。

表6 索玛鲁肽片剂PIONEER临床试验不良反应汇总
5.2 低血糖
索玛鲁肽片剂II期临床试验中有两例严重低血糖事件报告(索玛鲁肽注射剂组与索玛鲁肽片剂40 mg剂量快速增高组各1例),各组低血糖发作的总次数相似且较低[24]。索玛鲁肽片剂在肝肾功能损伤受试者的安全性研究中,索玛鲁肽片剂无严重低血糖事件的报告。7.0 %~8.0 %受试者出现无症状性低血糖(血糖浓度≤3.9 mmol/L或70 mg/dl),而仅有0 %~2.8 %的受试者被确诊为低血糖(<3.1 mmol/L或≤56 mg/dl)[15-17]。
5.3 其他
头痛为索玛鲁肽发生率较高的不良反应[15-17]。II期临床试验中有3例胰腺炎病例被确认[24]。在索玛鲁肽注射剂相关临床试验中也有急性胰腺炎病例的发生[37]。一名有腹膜炎病史的严重肝损伤受试者在临床试验期间死于细菌性腹膜炎;研究人员评估认为该事件与索玛鲁肽片剂相关性小[15]。有研究发现,索玛鲁肽可导致啮齿类动物甲状腺c细胞肿瘤的发病率以剂量相关、时间依赖的方式增加[37]。然而,在有关索玛鲁肽片剂的临床试验中尚未由甲状腺相关不良反应的报告。
6 总结
渗透增强剂SNAC在跨上皮细胞途径提高了索玛鲁肽表观渗透率,使得索玛鲁肽血浆暴露量增多,生物利用度提高。尽管此前SUSTAIN6已证实索玛鲁肽的心血管获益结果,但PIONEER6临床试验研究结果显示,索玛鲁肽片剂心血管安全性未能得到统计学上的支持。研究人员认为这一结果与试验持续时间过短有关。认为索玛鲁肽片剂口服给药可提高患者依从性,然而索玛鲁肽片剂的使用效果依赖于特殊的给药方式:服药前后需禁食一段时间。这一特殊给药方式的依从性需要患者的选择来证明。索玛鲁肽片剂在控制血糖,减少体重方面的有效性,为2型糖尿病患者提供了一个新的选择。
