阳极电致变色材料的研究进展
2019-12-06付子怡韦友秀刘伟明马一博李佳明李久勇
付子怡, 韦友秀, 刘伟明, 陈 牧, 马一博, 李佳明, 李久勇, 颜 悦
(1.中国航发北京航空材料研究院 透明件研究所,北京 100095;2.北京市先进运载系统结构透明件工程技术研究中心,北京 100095)
电致变色(electrochromic, EC)材料的特征是在外界电压驱动下,其光学性质由于发生氧化还原反应而改变[1-2],外观上表现为颜色发生变化,由电致变色材料组成的器件称为电致变色器件(electrochromic device,ECD)。通过电驱动改变光学性质的特性使得电致变色技术在很多领域得到应用,例如智能窗、汽车防眩后视镜、显示器等。图1 为电致变色技术的应用领域。
自20 世纪70 年代初,Deb 研制出第一个薄膜电致变色器件以来,有关电致变色技术的研究就从未停止过。美国的SAGE 公司首先开发了电致变色智能窗产品,是迄今为止报道的最合适的建筑变色窗,面积可达米级,在小于5 V 的直流电压驱动下即可变色,工作温度-30~60 ℃,循环寿命达105次。美国Gentex 公司是汽车防眩后视镜主要的生产商,产品已安装在了世界各地220 多款车型中。2005 年,Gentex 公司与PPG 航空工业公司合作在波音787 飞机中安装电致变色窗,乘客可以通过5 档按钮调节舷窗的亮度,使机舱变得更舒适。2008 年7 月,电致变色舷窗投入使用[3]。2017 年,日本旭硝子集团与美国Kinestral 科技公司联合新建了Halio 系列智能玻璃。国内相关研究起始于20 世纪90 年代,对电致变色器件的研究主要集中在高校和研究院中。国内电致变色企业相对于国外同类企业,存在着电致变色材料相对单一,缺乏自主产权等问题。浙江上方电子装备有限公司已实现智能玻璃的中试,2016 年研发出620 mm ×840 mm 的智能玻璃,已申请多项专利。合肥威迪制备出了全固态电致变色智能玻璃,能在-40~90 ℃环境中工作,响应时间约为2 min,可由透明态浅灰色至黑灰色(官网报道)。
典型的固态电致变色器件的结构如图2 所示,最外层是透明导电层[4-6],作用是将电子从外电路传输到变色材料中。中间是电解质层,主要用来导通离子,将电致变色层和离子储存层隔离开。电致变色层是核心层,是调制光学性质的主要材料。一般构成电致变色层的为阴极变色材料。离子储存层提供和储存离子,起到平衡电荷的作用,要求具有足够且稳定的电荷容量,一般构成离子存储层的为阳极变色材料。电致变色材料分为两大类:无机材料和有机材料,有机电致变色材料易进行分子设计、色彩丰富且变色速率快、吸收波长范围广,但其化学性质不稳定、水氧及紫外抗性较差、制备繁琐、成本高。无机电致变色材料变色颜色单一,变色速率慢,但其结构稳定,受空气中水和氧影响较小,且几乎不受太阳光紫外线影响。较好的耐候性使得无机电致变色材料更具有实际应用价值。

图 2 电致变色器件的经典结构Fig. 2 Typical structure illustration of ECD
电致变色层、电解质层和离子存储层的性质,以及三者的匹配性和组装状态都会影响器件的性能。目前商业化的无机电致变色器件并没有大面积的应用,存在响应速率慢、漏电流较大等问题。电解质和阳极电致变色材料的选择和制备是提高器件性能和实现应用化的关键,降低大面积薄膜和器件的制备成本也是该项技术普及化的重要因素之一。然而,目前缺少性质优异的阳极电致变色材料作为离子存储层应用在器件中。本文就近年来国内外有关阳极电致变色材料的研究进展做了概述,并对其今后的研究和发展提出一些建议。
1 阳极电致变色材料研究现状
1.1 无机阳极电致变色材料
无机电致变色材料主要包括过渡金属氧化物,按照着色的方式可以分为阴极和阳极着色材料,如W、Mo、Nb、Ta、Ti 等氧化物属于阴极着色材料。阳极电致变色材料主要是第VIII 族过渡族金属氧化物,如:Ni、Co、Mn、Ir 等氧化物及其水合氧化物,被还原时呈褪色态,被氧化时为着色态,普鲁士蓝系统也属于阳极着色材料。在氧化态和还原态下均有颜色,显示双着色性的材料如:V2O5、Rh2O3、CoOx也属于阳极着色材料。
(1)NiO
最典型的阳极材料是NiO[7],它是最常见的与WO3匹配的阳极材料。具有价格低廉、着色效率高、光调制范围大、循环稳定性较好等优点。它是一种P 型半导体材料,为立方型晶体,其电致变色效应主要体现在紫外和可见光区,在光学上属于中性色调,对透射光无附加颜色效应,在着色状态下具有柔和的中性颜色(灰色),接近于人眼对光波的敏感波段,在褪色状态下比较透明。与WO3薄膜相比较,NiO 电致变色薄膜电荷存储量小,而且稳定性由制备方法决定,有的制备方法不适合工业生产。
自从发现电致变色现象以来,研究者对变色机理的探索做了大量的工作,提出了一系列变色机理,常见的有3 种变色模型:色心模型、双注入模型和极化模型。这些模型在解释WO3的变色过程上都取得了一定的成功。而NiO 由于结构的致密性(NaCl 型结构),这些模型不能很好地解释NiO 的变色过程。至今NiO 薄膜的变色机理仍有很多争议。
在研究NiO 电致变色薄膜的过程中,采用的电解质多为KOH 等碱溶液。普遍被接受的反应机理是Bode 等[8]提出的,即变色主要是Ni(OH)2脱氢导致的,生成了含有Ni3+的NiOOH,如式(1)所示,但这个机理至今未能找到直接的证据证明。Carpenter等[9]则认为变色反应是由OH-的插入和抽出所产生的。Nemetz 等[10]将NiO 薄膜在着褪色状态下进行红外分析,发现在OH-伸缩振动的3400 cm-1位置,着色态薄膜的吸收大于褪色态薄膜,表明更多的OH-注入了薄膜。变色反应是因OH-的插入和抽出所产生的,反应方程式可以写为式(2)和式(3)。虽然争议很多,但是变色是由Ni2+变为Ni3+这一点是没有异议的。因此,NiO 的变色过程中可能发生如下反应[11]:

(2)V2O5
V2O5具有最高价态的矾,是最稳定的矾氧化合物,其晶体是斜方晶系单元晶胞结构,晶体参数是a = 1.151 nm,b = 0.356 nm,c = 0.437 nm。V2O5的结构可以想象为VO4四面体单元通过氧桥结合为边形成链状,具有典型的层状结构,有利于离子的嵌入和脱出,可应用于电致变色显示器件、锂离子电池和燃料电池电极等很多领域[12]。
V2O5是较为特殊的电致变色材料,具有多色的电致变色性能,当锂离子注入后,薄膜发生着色反应,在可见光区的透光率逐渐减小,而光谱短波范围内薄膜透光率有所升高(消色态),即V2O5在一个驱动电位下同时具有消色/着色态。其电致变色反应方程为:

非晶态的V2O5薄膜为黄色,在进行氧化还原反应时,可显示橙黄、绿色以及灰蓝色。在一定膜厚范围内,非晶态的V2O5氧化还原态的透过率相差不大,而且具有较高的电荷存储量,所以可以作为阳极电致变色材料使用。然而V2O5薄膜也具有很多不足,当其发生电化学反应时,会涉及大量锂离子嵌入和抽出的过程,可能会引起层状结构的塌缩或扭曲,引起结构破坏,带来的后果是电化学循环稳定性较差、薄膜电导率较低、锂离子扩散系数小,导致薄膜的着褪色对比度降低、着色效率变差、稳定性下降,这使得V2O5薄膜在电致变色的应用受到限制[13]。
(3)CeO2
Ce 的氧化物可以在还原态和氧化态之间转换(Ce3+↔Ce4+),在氧化还原过程中始终处于无色透明状态,插入/提取离子过程期间其透射率基本保持不变[14-15],在可见光范围内的透过率较高。
研究表明,每个Ce 可以吸收0.5 个Li 离子[16]。文献认为这很可能是由于Ce 氧化物在6 eV 宽的带隙中存在着窄的Ce4f能带[17]。
CeO2中的Ce 离子通常被认为是四价的,没有4f 态电子。而BIS 和XPS 显示带隙中存在部分空的4f 态,4d 跃迁到4f 的吸收光谱图也印证了这一点[18]。Koelling 等[19]的计算解释了XPS 和BIS 数据,他们发现在CeO2中,O2p价带和Ce5d导带的带隙宽度约为6 eV,在这个带隙内存在着窄的Ce4f能带,平均4f 层电子数约为0.5。这就解释了为什么每个Ce 可以吸收0.5 个Li 离子。
Ce3+不稳定,当施加外界电压,使Ce4+还原为Ce3+时,从晶格上失去相当数量的氧,形成大量氧空位(h),而所产生的氧空位又可将Ce3+氧化[20]:
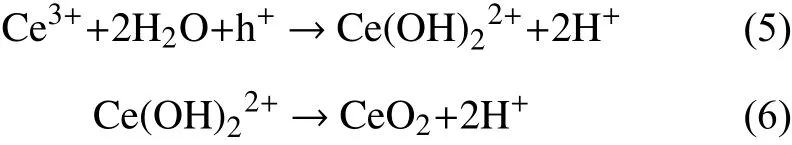
此时CeO2-x仍然能保持萤石型晶体结构,在外界电压、电流或者其他氧化还原刺激下都可以恢复之前的结构,因此CeO2的循环可逆性很好。然而,由于CeO2电荷量低,当它与WO3电极组成电致变色器件时,器件着色较浅,反应速率慢而且电化学性能不稳定,因此在电致变色智能窗中使用CeO2作为对电极是一个巨大的问题。
(4)IrOx
氧化铱(IrOx)也是一种常见的具有电致变色效应的材料,具有着色和褪色时间短、可逆性好和循环稳定性好等良好性能。其薄膜在施加电压后,可在透明态和蓝黑色之间进行颜色转换[21-22],IrOx的晶态和非晶态均有电致变色的特性。氧化铱的变色机理现在主要有两种,分别为H+的嵌入和OH-的抽出,如下式所示:

但价格昂贵、着色效率低、褪色状态有残余吸收等致命缺点,阻碍其成为理想的电致变色材料[23]。
(5)MnO2
二氧化锰(MnO2)具有多色的电致变色特性。锰在+4 价态时,由于d-d 跃迁,在可见光范围内具有光吸收,显示出棕色,Mn4+发生还原反应变为Mn3+时,颜色从棕色变为浅黄色,其电致变色反应方程为[24]:

二氧化锰作为阳极材料的主要缺点是其电荷储存量并不高,有待改性。而且褪色态为浅黄色,会影响器件褪色态的颜色。
(6)Co2O3
钴的氧化物可呈现三种不同的颜色:氧化亚钴(CoO,绿色)、氧化钴(Co2O3,蓝色)和四氧化三钴(Co3O4,褐色)。Burke 等[25]首先发现了钴的氧化物的电致变色现象。Co2O3薄膜可在蓝色和褐色之间可逆变化,而CoO 薄膜可在绿色和褐色之间可逆变化,其变化机理分别为:
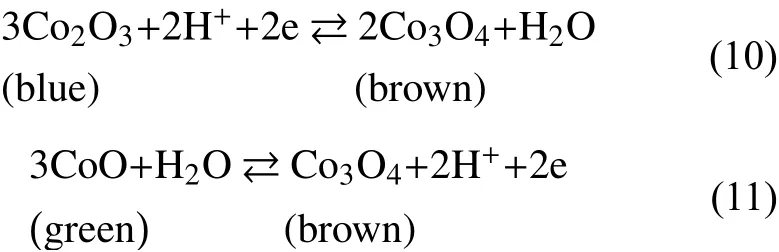
其中,反应式(10)的施加电压比反应式(11)的施加电压大。与其他阳极电致变色材料相比,氧化钴本身不稳定,颜色对比度小,循环稳定性差[26-27],在薄膜较厚的情况下,会发生钝化现象,只有部分钴元素参与氧化还原反应。
(7)普鲁士蓝(PB)
1978 年,Neff[28]首次发表了有关普鲁士蓝电致变色性能的文章。普鲁士蓝KFe[Fe(CN)6](Prussian blue,PB,)是混合价态的复合物,通式为MxI[MyII(CN)6],MI,MII为Fe 的两种价态的离子(Fe2+,Fe3+)。PB 是一个立方结构,按Fe2+―C≡N―Fe3+排列形成三维立方框架,Fe2+与Fe3+位于面心立方结构的顶点位置,由位于棱边上的―C≡N―连接,这样Fe3+被氮原子八面体包围,Fe2+被碳原子包围,立方体空隙的尺寸较大,能实现离子的快速迁移,因此近年来成为极有潜力的储能材料。在不同的氧化还原电压下,PB 可在普鲁士棕(PX),普鲁士绿(PG),普鲁士蓝(PB),普鲁士白(PW)之间转化,颜色在棕色,绿色,蓝色和无色之间进行转变,所以目前已经作为阳极变色材料应用在电致变色中。PX 是在PB 完全氧化下获得的,是不可逆的状态。实际应用中主要利用PB 蓝色和无色之间可逆转变,其特点是响应速率快,循环稳定性高,可达105次数量级[29]。氧化还原的过程如下[30]:
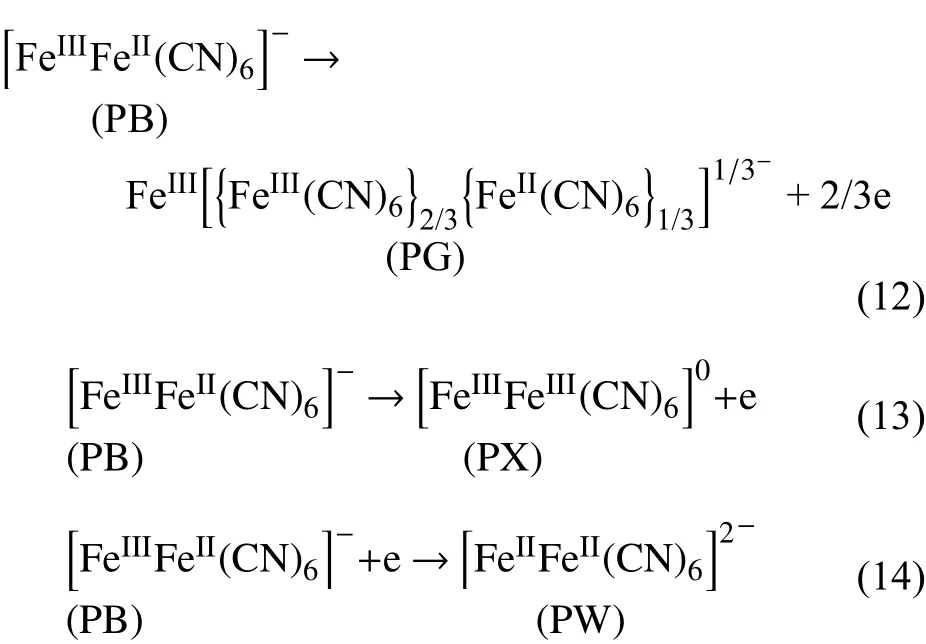
PB 与其他金属组成的复合材料(PBA)也具有各种颜色变化[31],例如,钴基PBA(Co-PBA,K2CoII[FeII(CN)6])可实现绿色与红棕色的颜色转换;铜基PBA(Cu-PBA,K2CuII[FeII(CN)6])可实现黄色与红色的颜色转换;镍基PBA(Ni-PBA,K2NiII[FeII(CN)6])可实现无色与黄色的颜色转换;钌基PBA(Ru-PBA)可实现无色与紫色的颜色转换;锌基PBA(ZnHCF)和铟基PBA(InHCF)在氧化和还原条件下都处于无色透明状态。因此,PB 和PBA 一直是许多研究人员的关注重点。此外,通过控制金属的价态,PBA 会显示出不同的性能(例如Co-Fe PBA 中的Fe 的价态[32]),使其应用更加广泛。PB 还可应用在电池、传感器、电催化和去除放射性元素等领域。
无机电致变色材料发展比较早,技术相对成熟,化学稳定性好,应用比较广泛,但无机电致变色材料具有颜色变化少,转变速率慢、电荷存储量相对于WO3薄膜较小和循环稳定性差等缺点。如何进一步提高现有无机阳极变色材料的性能也成为了人们的研究热点。
1.2 有机阳极材料
有机阳极电致变色材料主要分为聚合物和有机小分子两大类,聚合物有机电致变色材料有聚苯胺、聚毗咯、聚噻吩等,有机小分子电致变色材料包括吩嗪类、吡嗪类等[33]。有机材料具有着色效率高、响应速率快、颜色较为丰富、制备成本低、薄膜制备过程简单等优点,在对耐候性要求低的领域得到广泛的应用。常见的有机阳极电致变色材料有以下几种:
(1)吩嗪类
吩嗪,又名夹二氮杂蒽,是由一个吡嗪环左右两边分别连接一个苯环构成,结构如图3(a)所示,吡嗪环是它的发色基团,通过在氮原子的邻位上引入不同的基团,来实现颜色变化,在吩嗪上引入不同基团的化合物统称为吩嗪类衍生物,如图3(b)所示。根据苯环上引入的基团不同,吩嗪类化合物可以分为以下几类:(1)喹喔啉类,一般由邻苯二醌与邻苯二胺缩合而成,或者由邻苯二胺与邻二醛经环合而得,如由菲醌和N-苯基邻苯二胺缩合而得到的化合物;(2)氨基吩嗪和羟基吩嗪,如碱性红5;(3)单氨基间芳香吩嗪,如阿朴藏红;(4)藏红,如碱性紫、藏花红T;(5)引杜林,如溶剂蓝7[34]。

图 3 吩嗪结构 (a)和吩嗪类衍生物基本结构(b)Fig. 3 Structure of phenazine (a)and basic structure of phenazine derivatives(b)
吩嗪类化合物在还原态时没有颜色,被称为褪色态,被空气或者其他氧化物氧化之后,会变成各种颜色。一般来说,要想得到红色、橙色和品红等色调,在吩嗪化合物的3-位或者7-位或者3,7-位基本都要有未被取代的氨基。但是当3-位或者7-位上有氨基时,化合物在含有硝酸盐的体系中不稳定,因此Miller 等[35]提出将具有吸电子取代基的酰基引入到10-位,酰基与氨基共同作用可以加强吩嗪类化合物的稳定性。
(2)导电聚合物
导电聚合物又称共轭聚合物,是由各种有机芳香分子利用化学或电化学的方法合成的,通过改变施加在导电聚合物上的电压,可以改变其掺杂程度,也就改变了其能隙Eg的大小,从而改变其吸收光波长,从视觉上表现为颜色的变化[36]。掺杂的浓度不同可以使一些导电聚合物在多种颜色之间转换。在外电压驱动下,导电聚合物在高电位区会发生P 型掺杂/脱掺杂(氧化态/中性态)反应,在低电位区又会发生N 型掺杂/脱掺杂(还原态/中性态)反应,并且其电导率随着掺杂/脱掺杂会发生改变,掺杂后电导率增加,脱掺杂后电导率降低。大部分的共轭聚合物是P 型掺杂变色,即聚合物在中性态和氧化态之间转换。
阳极导电聚合物材料本征态为高带隙聚合物,光吸收主要位于紫外区,所以在不施加外电压,或者施加负电压时,材料呈褪色态;在施加正电压时,掺杂度增大,带隙变窄,光吸收移入可见光区,材料呈着色态[37]。
聚苯胺(PANI)是一种比较典型的阳极导电聚合物,在氧化还原过程中可稳定呈现出多种颜色,在完全还原状态下显示淡黄色,施加一定电压后,在部分氧化状态下显示出绿色或者蓝色,再继续施加电压,在全氧化状态下显示紫色,但是此时其氧化还原反应将不具有可逆性,表明此时施加电压过高。也就是说,施加的电压在一定范围内,聚苯胺的颜色可以在淡黄色、绿色和蓝色之间可逆转变,且循环次数可达百万次以上,可是超过了这一电压范围使其氧化成了紫色,聚苯胺的稳定性和循环可逆性将急剧下降[38-40]。
聚吡咯(PPy)的变色原理与聚苯胺相似,也是在不同电压下掺杂程度不同引起带隙变化,导致光吸收变化,视觉上显示出颜色变化。加负电压时,聚吡咯为完全脱掺杂状态,此时的聚吡咯几乎不导电,薄膜呈淡黄色,电压变化到0 V 时,薄膜呈部分掺杂态,颜色加深变为绿色,继续施加正向电压,双极化子能带形成,并在700 nm 处形成吸收,此时薄膜呈现蓝色,当电压施加过高到1 V,聚吡咯进入高掺杂状态,很可能过度掺杂,此时薄膜呈现紫色,其可逆性被部分破坏,难以回到完全脱掺杂态。聚吡咯合成简便,原料便宜,但是其吸收系数大,因此底色较强,需要将其制备成很薄的薄膜,才能清楚地看出其还原态和氧化态的颜色变化[41-43]。
导电聚合物有许多优于无机化合物的特点,比如高着色效率,快速感应能力,同一种材料有着丰富的颜色,通过改变化学结构可获得好的能带转变。与无机电致变色性材料相比,导电聚合物着色效率高,响应能力快,而且同一种材料能显示出多种颜色,在加工性、制备方法和费用等方面具有明显的优势,然而其循环稳定性和耐候性较差,限制了其实际应用。
2 变色薄膜制备方法
电致变色材料在器件中通常以薄膜形式存在,如何制备大面积均匀薄膜是实现该技术工业化首要解决的技术问题之一。制备薄膜方法主要有三大类:物理气相沉积(PVD)[44-46]、化学气相沉积(CVD)[47-48]、基于溶液的湿化学法[49-53]。PVD 和CVD 主要用来制备无机电致变色材料薄膜,成膜均匀,纯度高;湿化学法制备的电致变色材料薄膜种类较多,并且能制备多元复合材料,成本低,不需要昂贵的实验设备。
2.1 物理气相沉积(PVD)
PVD 技术主要的工作原理是采用物理方法,将材料表面气化成气态原子、分子或部分电离成离子,并通过系统中的低压气体或是等离子体,在基底表面沉积得到具有某种特殊功能的薄膜。PVD的主要方法包括:磁控溅射、离子镀、脉冲激光沉积等。图4 为两种物理气相沉积法磁控溅射和离子镀原理示意图。

图 4 物理气相沉积原理示意图 (a)磁控溅射;(b)离子镀Fig. 4 Schematic diagram of PVD (a)magnetron sputtering;(b)ion plating
这些方法主要是由真空蒸发演变而来的,真空蒸发是物理气相沉积法中使用最早的技术,然而实验条件限制了使用此方法的原材料的种类,多用来沉积金属或金属化合物。
磁控溅射是制备过渡金属氧化物薄膜最常用的技术[54],原理如图4(a)所示,利用磁场与电场交互作用,使电子在靶表面附近成螺旋状运行,与氩气撞击产生带电离子,所产生的带电离子在电场作用下撞向靶面从而溅射出靶材。此方法的优点是沉积速率快,薄膜与基片结合较好,纯度高,致密性好,成膜均匀。但其靶材的利用率低,不适合制备多元掺杂的薄膜,而且不能实现强磁性材料的低温高速溅射。
张泽华等[55]采用直流磁控溅射法,使用纯度为99.99%的高纯Ni 靶,在FTO 薄膜上沉积了不同厚度的NiO 薄膜。结果表明,厚度为920 nm 的薄膜着色效率最高,达到了23.46 cm2/C,而厚度为80 nm 的试样光学调制幅度最大,着色、褪色时间最快,在波长为550 nm 处,光学调制幅度为40.85%,着褪色响应时间分别为4.47 s 和2.28 s。
Wei 等[56]采用磁控溅射法分别制备了WO3和NiO 薄膜。采用的W 靶纯度为99.5%,Ni 靶纯度为99.9%,基底为ITO 导电玻璃,通过控制O2/Ar气体流量比和沉积时间来控制NiO 薄膜的厚度。结果表明,O2/Ar 气体流量比控制在1.7/23.3,沉积时间为75 min 时,得到的厚度为540 nm 的NiO 薄膜性能最优异,光学调制幅度大,电荷密度可达10.22 mC/cm2。
离子镀是在真空条件下,利用气体放电使气体或被蒸发物部分离子化,最终将蒸发物或反应物沉积在基片上,是结合蒸发与溅射两种薄膜沉积技术而发展的一种 PVD 方法,原理如图4(b)所示。其优点是所镀薄膜与基片接触良好,可在低温衬底沉积,避免高温引起的扩散。
Niwa 等[57]使用射频离子镀的方法,以ITO 为透明导电层,制备了锡掺杂氧化铱的薄膜(IRTOF),薄膜中如果Ir 的含量小则着色效率低且制备时间较长,所以选择铱元素和锡元素的比为0.17∶0.83来制备薄膜并组装成电致变色器件,器件着褪色响应时间较短,透过率的可调控范围在15%~60%,并在10 万次循环后性能保持良好,且通过了耐热性实验。
李筱琳等[58]采用低压反应离子镀工艺制备了NiO 薄膜。将普通玻璃、ITO 玻璃和ITO 塑料等不同衬底同时放入真空室进行镀膜,保持温度室温,背底真空2 × 10-3Pa, 工作气体是氩气,氩分压为2.5 × 10-2Pa, 反应气体为氧气,采用不同的氧分压(分别为4 × 10-2Pa、5 × 10-2Pa、6 × 10-2Pa、7 ×10-2Pa)镀膜,电子枪功率6 kW,束流0.08 A,离子流7~10 A。沉积速率监控在1~2 nm/s。在不同氧分压条件下,控制镀膜时间,使其膜厚在280 nm左右。结果表明,当氧分压为6 × 10-2Pa 时,制备的NiO 薄膜具有较为优越的电致变色特性。
2.2 化学气相沉积(CVD)
CVD 是另一类获得薄膜的技术手段,如图5所示,利用气态或者蒸汽态的物质在气相或者气固界面上反应而生成固态沉积物。主要通过控制前驱体溶液来控制薄膜的成分。这种工艺技术沉积反应速率快,成分可调,已经广泛用于提纯物质、研制和制备各种晶体或玻璃态无机薄膜材料。目前,CVD 技术已成为无机化学的一个新领域。

图 5 化学气相沉积基本装置Fig. 5 Schematic diagram of CVD
Drosos 等[59]用气溶胶辅助化学气相沉积(AACVD)技术,在FTO 基底上沉积得到亮黄色的透明V2O5薄膜。该薄膜性能良好,在2000 次循环后性能基本保持不变,具有良好的结构稳定性和高可逆性。
Jahan 等[60]用CVD 法制备了聚(3-甲基噻吩)(P3MT)薄膜和聚(3-乙基噻吩)(P3HT)薄膜,在CVD 室中持续2 h,温度保持在60 ℃。最后得到的P3MT 可从深绿色变为深红色,P3HT 可从黄绿色变为红色。SEM 显示表面形成了平均尺寸为60 nm的均匀纳米颗粒,EIS 表明P3HT 涂层聚酯具有更高的导电性。
2.3 湿化学法
20 世纪90 年代,出现了各种各样利用溶胶凝胶(sol-gel)、喷雾、电沉积、旋涂等化学方法制备无机电致变色材料薄膜的报道,此类统称为湿化学法,湿化学法制备无机电致变色薄膜和器件逐渐成为一个日益活跃的研究领域。
旋涂法是在平坦的基板上制备均匀薄膜的方法,主要过程是将溶液滴在基板上,然后利用旋涂仪旋转基板,溶液在离心力的作用下平铺在基板上,最后成膜。主要通过调节转速来控制薄膜的厚度,一般来讲转速越快制备的薄膜越薄,其次也跟溶液的黏度和浓度有关系。这种方法在制备小面积的薄膜的时候很便利,制备大面积的薄膜的时候,随着半径的扩大,离心力逐渐增大会导致薄膜的厚度由里向外逐渐递减,不适合产业化生产。
Wei 等[61]使用旋涂法制备了Ti 掺杂V2O5薄膜。将VO(OC3H7)3溶液Ti(OC3H7)4溶液分别溶解在异丙醇,然后以2∶1 的体积比混合,制备V∶Ti=2∶1 的涂布溶液。在1000 r/min 的速率下每次旋涂30 s,获得厚度约为40 nm 的Ti 掺杂V2O5膜。通过增加旋涂次数增加膜厚度。将涂层在300 ℃温度下退火30 min 以除去溶剂。结果表明,厚度为100 nm 左右的薄膜性能最优异,薄膜在保持较好的电荷量的同时,在氧化和还原状态下具有较小的光学调制幅度,可用作离子存储层。
Liao 等[62]用旋涂法制备了PB/PEDOT:PSS 薄膜,旋涂液由PB 前驱体溶液和透明导电PEDOT:PSS溶液混合而成。然后,将涂层溶液以700 r/min 的转速旋涂到ITO 基板上。最后,将其在130 ℃下固化,制备出了PB/PEDOT∶PSS 复合薄膜,可在无色(ES)、蓝色(PB)、绿色(BG)和黄色(PY)之间转换,最高光学调控幅度为80%,如图6所示。将其与MoOHCF 组装成电致变色器件,可实现深绿色和黄色之间的颜色转变,对于大多数可见波长,光学调制范围约为40%。
Noonuruk 等[63]使用旋涂法,在FTO 衬底上制备了Zn 掺杂的NiO 薄膜。通过控制加入前驱体溶液中乙酸锌二水合物(Zn(CH3COO)2•2H2O)的体积,得到不同掺杂比例的前驱体溶液,从而得到不同掺杂比例的薄膜。结果表明,Zn 的掺杂会降低NiO 薄膜的结晶度,从而扩大薄膜表面活性表面积,便于电荷的插入和抽出。掺杂Zn 的浓度为10%时,薄膜电化学性能最优异,此时光学调制幅度最大,CV 曲线中阳极和阴极电流峰值最高,表明Zn 掺杂的NiO 薄膜在发生氧化还原反应过程中被注入/抽出了更多的电荷,电致变色性能更好,薄膜交换的锂离子量增多。

图 6 PB/PEDOT:PSS 薄膜[62] (a)循环伏安图;(b)原位平衡透射光谱图Fig. 6 PB/PEDOT:PSS film[62] (a)cyclic voltammogram;(b)in situ equilibrated transmittance spectra
喷雾热解主要的原理是先以水、乙醇或其他溶剂将反应原料配成溶液,将前驱体溶液与一定的气流混合,在强气流压的作用下从喷雾嘴喷出雾气,喷出的雾气沉淀在加热的基板上发生热解成膜[64]。通过多次的加压喷雾来增加薄膜的厚度,对基底的表面形状没有要求。这种方法可以很方便地制备多元合金氧化物材料,主要通过控制前驱体溶液的成分和比例来获得多元合金氧化物材料。
Elshorbagy 等[65]用自制喷雾热解法制备了普鲁士蓝薄膜。通过控制喷嘴到基板的距离、基板表面温度、溶液流速、沉积时间和沉积速率这五个参数来制备PB 薄膜。性能最优异的PB 膜形貌良好,表面光滑,着色效率高,循环过程中电化学性能良好,为将来利用掺杂剂开发薄膜性能开辟了新的道路。
Denayer 等[66]采用超声喷雾热解法(USP)分别将掺杂锂的NiO(LiNiO)和未掺杂的NiO 薄膜沉积在FTO 基底上,并通过在前驱体溶液中加入聚乙二醇(PEG),研究这种表面活性剂辅助USP 技术对NiO 和LiNiO 薄膜形成的影响。结果表明,与不含表面活性剂的薄膜相比,在前驱体溶液中加入PEG 可以改善均匀性并减少光散射,锂离子的存在会改善NiO 薄膜的电致变色性能,因此,LiNiO-PEG 薄膜的性能最优异,光学调制范围为43.5%,着色效率达到了41.2 cm2/C。
水热合成法是指把反应前驱液转移到高压反应釜中,在一定温度和压强下,反应物之间进行反应生成产物,图7(a)为水热反应釜的基本结构。水热合成的主要优点是在无任何结构控制剂或模板存在的条件下,通过选择适当的反应温度、时间、填充率以及所用溶剂等,能够较方便地合成各种纳米结构的产品。
Qian 等[67]用水热法制备了纳米结构的PB薄膜。在室温下将0.25 g 葡萄糖(C6H12O6)和0.66 g铁氰化钾溶于60.0 mL 去离子水中磁力搅拌,然后将1.0 mL 浓盐酸滴入溶液中调节溶液PH 值,在不锈钢高压釜中水热合成PB 纳米薄膜。所制备的PB 纳米薄膜与基板具有很好的黏合性,且具有大量纳米通道能转移离子,使反应能够迅速发生。将PB 膜组装成的电致变色器件性能良好,具有良好的循环稳定性,显示出2.4 s 的着色响应和1.0 s 褪色响应以及87.4 cm2/C 的高着色效率。
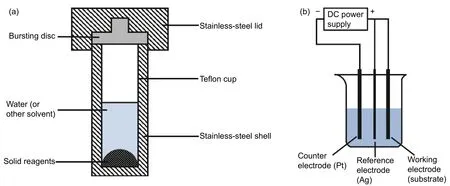
图 7 水热合成法 (a)反应釜;(b)三电极装置Fig. 7 Hydrothermal synthesis (a)reactor;(b)three-electrode device
Chu 等[68]采用简单的一步水热法制备了纳米棒状V2O5。在FTO 基底上,使用NH4VO3和草酸作为前驱体溶液,将其放入不锈钢高压釜中,在180 ℃下水热3.5 h,冷却过夜后将样品在300 ℃下退火1 h,然后在500 ℃下退火2 h 得到V2O5薄膜。通过分别施加 ± 0.5 V 的电势,可目视观察到V2O5在着褪色时呈现黄色到淡蓝色的颜色变化。
Cao 等[69]采用水热合成法制备了平均直径约为10 nm 的NiO 薄膜。使用Ni(NO3)2和HMT(六亚甲基四胺)制备出前驱体溶液,然后将其转移到不锈钢高压釜中,将2.5 cm × 2.5 cm 的ITO 玻璃浸入反应溶液中,然后将高压釜密封并在105 ℃下保持1 h,并使其自然冷却至室温。反应后,取出基质,用去离子水完全洗涤,并在空气中干燥。最后,将样品在300 ℃下,在氩气中退火1.5 h,形成多孔NiO 纳米壁阵列。与致密的NiO 膜相比,多孔NiO 纳米壁阵列表现出较弱的偏振,较高的色彩对比度、较好的反应性和循环性能。中孔NiO 纳米壁阵列(纳米粒子的距离约为5~20 nm)表现出优异的电致变色性能,在波长为550 nm 处,透射率变化高达77%,并且计算出其着色效率为49 cm2/C。循环稳定性也良好。
电沉积技术制备电致变色材料薄膜的主要过程就是在前驱体溶胶或溶液中施加合适的电场,溶胶或溶液中的有效带电成分在电场强度的作用下移动到基板上,并在基板上发生化学反应最后成膜[70],一般采用如图7(b)所示的三电极装置。该技术的重点是选择合适的电镀液,其成分和pH 值会大大影响薄膜的成分和形貌。与上述多种湿化学法相比较,电化学沉积技术工艺简单、设备投资少、容易操作、生产方式比较灵活、是一种经济的沉积技术。
Assis 等[71]使用恒电流电沉积法制备了普鲁士蓝纳米薄膜。将相同浓度的稀盐酸(HCl)、铁氰化钾(K3Fe(CN)6)、六水合三氯化铁(FeCl3•6H2O)按1∶2∶2 的比例混合,在-40 μA/cm2的恒电流下沉积300 s 得到厚度为217 nm 的普鲁士蓝薄膜。以普鲁士蓝薄膜组装得到的电致变色器件透过率的差别在(35 ± 2)%,循环稳定性能一般,可能由于电解质层是固态,离子传输能力受到影响。
Huang 等[72]采用电沉积法在ITO 玻璃上沉积了PB 薄膜和PEDOT 薄膜,其有效面积为2 cm × 2 cm,分别以这两个薄膜为阳极和阴极,以CSIC-PVB 作为电解质,制备出PB/CSIC-PVB/PEDOT 的电致变色器件,器件的最大光学调制范围为39.8%。着褪色响应时间分别为1.2 s 和1.5 s,计算出着色效率为270.8 cm2/C。
Bodurov 等[73]采用一步电沉积法在ITO 基底上制备NiO 薄膜。结果表明,电沉积法可以得到性能良好的NiO 薄膜,高温退火前,NiO 薄膜具有50%~60%的初始透过率,在400 ℃高温退火后,初始状态的透过率可达到90%。
溶胶凝胶法是以无机盐或金属醇盐为前驱体,经过水解缩聚过程逐渐凝胶化,然后作相应的后处理得到粉体材料,图8 为溶胶凝胶法制备薄膜的流程图。使用溶胶凝胶法能够得到高均一、高比表面积的薄膜,且能够较容易地控制材料的组成。
图 8 溶胶凝胶法制备薄膜的流程图Fig. 8 Flow chart of film preparation by sol-gel method
Falahatgar 等[74]采用溶胶凝胶法在ITO 基底上沉积得到了MnO2-ZnO 薄膜。通过控制前驱体溶液中Zn 的浓度得到不同摩尔比的MnO2-ZnO 薄膜,研究了Zn 含量的变化对非晶态MnO2-ZnO 薄膜电致变色性能的影响。AFM 的结果表明,薄膜表面粗糙度随Zn 浓度的增加而增加,在扫描速率为20 mv/s 时,摩尔比为25%的MnO2-ZnO 薄膜阳极和阴极的电流峰值最高,表明同样的电压范围内,此薄膜在发生氧化还原反应过程中被注入/抽出了更多的电荷,电致变色性能更好。
Zhou 等[75]采用溶胶凝胶法制备了NiO,Li-NiO,Ti-NiO 和Li-Ti-NiO 薄膜,为了 研 究Li 和Ti 的掺杂对NiO 薄膜性能的影响。将相应的试剂溶 解 在 乙 醇 中,Ni∶Li 和Ni∶Ti的摩 尔 比 分 别 为2∶1 和4∶1。结果表明,Li 和Ti 的掺杂会影响NiO立方相的对称性,增加晶格缺陷,能调节薄膜中Ni2+/Ni3+的比例,从而使得Li-Ti-NiO 薄膜的光学调制幅度更大。
Wu 等[76]采用溶胶-凝胶法合成了CeO2-TiO2薄膜。前驱体溶胶由硝酸铈六水合物Ce(NO3)3•6H2O,钛酸丁酯Ti(OC4H9)4和无水乙醇CH3CH2OH的混合物组成。使用溶胶凝胶法沉积膜并在氧气气氛中煅烧,温度为310 ℃。通过控制前驱体溶液的配比,研究了不同Ce、Ti 比(0.55∶1,0.70∶1,0.85∶1和1∶1)对(CeO2)x-TiO2薄膜微观结构和性能的影响。结果表明,Ce∶Ti=0.85∶1 时电荷密度最高,可达19.99 mC/cm2,且在着色和褪色状态下透过率变化很小,表明其可以用作离子存储层。
综上所述,湿化学法是一种工艺简单,成本低,可调控的制膜方法,但是湿化学方法在制备过程中受溶液或溶胶黏度、浓度梯度以及环境的影响较大,因此如何利用湿化学方法制备和生产均匀的大尺寸薄膜成为工业化应用的关键。
3 阳极变色材料的性能优化
电致变色技术虽然经历了几十年的研究,但是其产业化的道路还受到诸多限制,而且不同的材料和器件的问题也不尽相同。电致变色材料在器件中一般以薄膜的形式存在,变色过程会涉及大量锂离子嵌入和抽出的氧化还原反应,这会对薄膜材料的结构产生严重的影响,降低材料的变色性质会导致组装的器件的稳定性下降,而且阳极电致变色材料还存在电荷储存量低的问题。为了进一步提高其变色性质,学者们采用多种方法来对薄膜进行改性。
3.1 掺杂改性
掺杂是比较常见的材料改性方法,例如可以通过掺杂降低电致变色材料禁带宽度和结晶度,从而提高材料本身的导电性来提高材料的稳定性、电化学反应速率以及光学性质。根据要达到的效果有目的地选择掺杂元素和方法。一元掺杂比较常见,掺杂方法一般为磁控溅射和湿化学法。例如Venkatesh 等[27]用喷雾热解法制备了锰(Mn)掺杂的Co3O4,将其与未掺杂的Co3O4薄膜进行对比,掺杂Mn 浓度为6%的薄膜表现出优异的光学性能,如图9 所示,着色和褪色状态下的透过率调制范围可达35%。这是由于Mn 离子掺杂到Co3O4之间,产生局部晶格畸变,晶粒尺寸随着Mn 掺杂量的增加而减小,Mn 掺杂为6%时晶粒尺寸最小,此时变色材料可与电解质表面充分接触,从而提高了材料的电致变色性能。此外,掺杂的薄膜表面均匀地分布着多孔枝晶状结构,这种结构有利于离子的嵌入和抽出,从而提高薄膜的氧化还原反应速率,进一步提高薄膜的电致变色性能。

图 9 Co3O4 薄膜透过率曲线[27] (a)纯Co3O4;(b)掺杂4%(原子分数,下同)Mn;(c)掺杂6%Mn;(d)掺杂8%MnFig. 9 Transmission spectra of bleached and colored states of Co3O4 films[27] (a)pristine Co3O4;(b)Mn 4%(atom fraction,the same below)-doped;(c)Mn 6% -doped;(d)Mn 8% -doped
为了达到多重效果,还可以进行多元掺杂,一般采用湿化学法制备。Li 等[77]用溶胶凝胶法制备了介孔ZrO2-CeO2-TiO2多元掺杂薄膜,其SEM 图如图10 所示。Zr 的掺杂稳定了薄膜的长程有序多孔结构,而且比表面积大,使介孔ZrO2- CeO2-TiO2多元掺杂薄膜具有良好的热稳定性,Ce 的掺杂可以产生更多的氧空位,方便锂离子的嵌入和抽出。Zhou 等[75]用溶胶凝胶法制备了Li-Ti 共掺杂的NiO 薄膜(Li-Ti-NiO 薄膜),并制备了纯NiO 薄膜、Li 和Ti 离子单元掺杂的NiO 薄膜与其进行对比。实验结果表明,共掺杂的薄膜表现出比相应的单元离子掺杂更加优异的电致变色性能和稳定性。在共掺杂薄膜中,Li+和Ti+进入NiO 立方晶的晶格内,并取代部分Ni 位点,改变Ni2+/Ni3+的比例,扭曲NiO 立方晶晶体结构,降低结晶度,有效的提高NiO 薄膜的电致变色性能和循环稳定性。

图 10 TEM 图像[77] (a)TiO2;(b)CeO2-TiO2;(c)介孔CeO2-TiO2;(d)介孔ZrO2-CeO2-TiO2Fig. 10 TEM images[77] (a)TiO2;(b) CeO2-TiO2;(c)mesoporous CeO2-TiO2;(d) mesoporous ZrO2-CeO2-TiO2
除了掺杂,复合材料的体系也可改进阳极变色材料的性能。岳言芳等[78]先用水热法制备了NiO 薄膜,然后用电沉积法将PB 沉积到NiO 薄膜上,制得NiO/PB 复合电致变色薄膜,实验表明,复合薄膜具有更好的电致变色性能,其数据如表1 所示。NiO 和PB 同为阳极变色材料,因此,NiO/PB复合能一定程度上提高材料的电致变色性能,在波长为700 nm 处透过率差可达46%,着色效率高达121 cm2/C,着褪色响应时间分别为5 s 和6 s。

表 1 NiO 和NiO/PB 的响应时间和对比度比较Table 1 Switching time and contrast comparison of NiO andNiO/PB
3.2 纳米结构改性
另一改性方法是将电致变色材料纳米化,然后制备成薄膜。纳米结构的电致变色材料在至少一个维度上具有极短的电荷传输距离,而且具有极大的比表面积,可与电解质充分接触,因此电荷传输速率很快且电化学活性更高,从而可以显著增强电致变色性质。一般采用湿化学法来制备具有纳米结构的电致变色薄膜。

图 11 PANI 的SEM 照 片[79] (a)GO-ITO 上PANI 的 横 截 面 图;(b)ITO 上PANI 的 顶 视 图;(c)GO-ITO 上PANI 的顶视图Fig. 11 SEM images of PANI[79] (a)cross-sectional view of PANI coated with GO-ITO;(b)top-view of PANI coated with ITO;(c)top-view of PANI coated with GO-ITO
纳米结构的电致变色材料主要是通过两种方式得到。第一种方式是在原本的电致变色薄膜上引入导电纳米材料,例如碳纳米管(CNT)、石墨烯、ITO 纳米颗粒等来促进电致变色材料中的电子传导和离子传输。吕晓静等[79]先在ITO 上制备了氧化石墨烯(GO)薄膜,然后分别在ITO 电极和GO-ITO 复合电极上制备了PANI。图11 为复合纳米薄膜的SEM 照片,可以看出,界面之间紧密结合,证明GO 与ITO 及聚合物薄膜之间具有良好的界面相互作用。在GO-ITO 电极上制备的PANI 颗粒粒径更小,表面也更加均匀平整,从而增大了聚合物与电解液之间的接触面积,为电致变色过程中离子的插入和抽出提供了更多的通道,提高了材料的电致变色性能。光学和电学测试结果表明,GO 的引入确实使PANI薄膜的光学对比度在波长为700 nm 处提高了约13%,着色效率高达169.6 cm2/C,响应时间也缩短了约2.6 s。Nossol 等[80]在ITO 基板上分别制备了PB/碳纳米管(PB/CNT)复合膜和PB 单层膜,并比较了它们的电致变色性能。结果表明,引入CNT 之后,薄膜的循环稳定性得到明显提升,500 次循环后的最大透过率差仍能保持第一次循环的90%。
第二种方式是将电致变色材料制备成纳米结构,例如制备成纳米管、纳米棒、纳米微粒等,其结构松散孔隙大,离子的运动更加容易。Cai 等[81]用水热法和化学浴沉积法制备了TiO2/NiO 纳米棒阵列薄膜,并与NiO 纳米薄膜进行对比,发现TiO2/NiO纳米棒阵列薄膜具有更好的电致变色性能,图12为TiO2纳米棒阵列、TiO2/NiO 纳米棒阵列和NiO纳米薄膜的SEM 图像。从图12 可以看出,在沉积NiO 之前,FTO 基板的整个表面均匀地覆盖着具有矩形横截面的TiO2纳米棒,在涂覆NiO 之后,多孔网状的NiO 纳米薄片均匀地覆盖在TiO2纳米棒的整个表面,并且TiO2纳米棒的排列保持不变。TiO2纳米棒状结构和NiO 的超薄层状多孔结构起到了很好的协同作用,可促进离子的扩散并增加电子的传输速率,提供了更大的比表面积,可使变色材料与电解质表面充分接触,从而提高了材料的电致变色性能,数据对比如表2 所示。

图 12 SEM 图像 (a,d)TiO2 纳米棒阵列;(b,e)TiO2/NiO 纳米棒阵列;(c,f)NiO 纳米薄膜Fig. 12 SEM images (a,d)TiO2 nanorod array;(b,e)TiO2/NiO core/shell nanorod array;(c,f)NiO nanoflake film

表 2 NiO 纳米薄膜和TiO2/NiO 纳米棒阵列薄膜的响应时间和对比度比较Table 2 Switching time and contrast comparison of NiO nanoflake film and TiO2/NiO core/shell nanorod array film
Li 等[82]以液相沉积法先制得ZnO 纳米棒为模板,通过水解NiCl2得到了NiO 纳米棒。制得的NiO 纳米棒光学调制率高,在550 nm 处为78.5%,着褪色的响应时间快,分别为3.92 s 和3.43 s,电荷量高且循环稳定性好,电致变色性能十分优异。
4 未来展望
将电致变色技术转化成产品并且达到实际应用的技术指标是重要的发展趋势。阳极电致变色材料作为器件中的离子存储层,提高其循环稳定性和光学特性是提高器件性能实现应用的关键技术之一。而开发简单易行且成本低廉的薄膜制备工艺,也是电致变色技术应用化的研究重点。目前应用最广泛的阳极电致变色材料是NiO,其他无机阳极电致变色材料本身电化学性能不够优异,通过掺杂改性后可提高其性能,从而得到应用。有机阳极电致变色材料具有多种颜色变化,且变色速率快,但是其循环稳定性较差,是限制这类材料实际应用的主要原因。未来,还有很多新的阳极电致变色材料有待开发。例如,锂离子电池中的正极材料锂锰氧化物稳定性好,无污染,成本低廉;锂钴氧化物具有良好的电化学性能且循环性好;金属氧化物RuO2具有较高的电导率、良好的导电性和化学稳定性,被应用于超级电容器材料中。这些新材料有着优异的电荷存储能力,可用于制备器件中的离子存储层,但能否应用到电致变色中,取决于其光学性能是否能够满足要求,以及制备方法是否适用于大规模生产。
