高纯度庆大霉素C1a游离碱制备工艺研究
2019-12-03杨涛陈舟舟王海东李亚军李继安雷翔霄陈代杰储消和
杨涛 陈舟舟 王海东 李亚军 李继安 雷翔霄 陈代杰 储消和
(1 上海交通大学药学院,上海 200240;2 浙江工业大学长三角绿色协同创新中心,杭州 310014;3 常州方圆制药有限公司,常州 2130125;4 上海医药工业研究院创新药物与制药工艺国家重点实验室,上海 201203)
氨基糖苷类药物自1944年面世以来,其具有水溶性好、化学性质稳定、抗菌谱广、抗菌能力强和吸收排泄良好等特点,是治疗革兰阴性菌和结核杆菌感染的首选药物之一[1]。1963年美国Schering公司的Weinstein第一次从棘孢小单孢菌中分离得到庆大霉素(gentamicin)[2]。广义上的庆大霉素是多组分的混合物,以C族复合物为主;主要从绛红小单孢菌、棘孢小单孢菌中发酵分离获得,其主要组分有C1,C1a,C2,C2a。其中以C1a和C2a的治疗效果最好,但是易产生耐药性;C1和C2治疗效果一般,但是不易产生耐药性[3]。
其中庆大霉素C1a单组分是我国一类新药硫酸依替米星半合成的关键起始物料[4],而市售的庆大霉素C1a游离碱一般HPLC纯度都低于93%,严重影响依替米星质量和生产过程杂质控制。现有庆大霉素C1a生产工艺主要采用离子交换树脂静态和柱层析吸附-氨水解析法分离纯化庆大霉素生产过程富集的庆大霉素C1a粗品(HPLC纯度50%~60%)[5-6],但其难以获得高纯度的庆大霉素C1a。
本研究团队探索用大孔吸附树脂层析法及盐析结晶法精制纯化硫酸依替米星起始物料—庆大霉素C1a成品(HPLC纯度91%~93%),获得高质量的庆大霉素C1a精制品。最终希望通过减少庆大霉素C1a中杂质来减少依替米星中杂质水平,获得高质量的硫酸依替米星API产品。
1 材料和方法
1.1 仪器和材料
1.1.1 材料
本实验所用材料见表1。
1.1.2 仪器
本实验所用仪器见表2。
1.2 方法
1.2.1 庆大霉素C1a的含量及纯度测定
(1)庆大霉素C1a的含量测定:精密称取本品适量,用水定量稀释制成约含庆大霉素C1a 0.4mg/mL的溶液,作为供试品溶液;另取庆大霉素C1a对照品适量,精密称定,加水溶解并定量稀释制成约含庆大霉素C1a 2.0mg/mL的溶液,作为对照溶液;精密量取对照溶液适量,用水定量稀释制成含庆大霉素C1a各0.3、0.4和0.5mg/mL的溶液作为对照溶液(1)、(2)和(3)。照高效液相色谱法(中国药典2015年版四部·通则0521)试验,用十八烷基硅烷键合硅胶为填充剂(pH范围0.8~8.0);以0.2mol/L三氟醋酸溶液-甲醇(84:16)为流动相;流速为0.5mL/min;用蒸发光散射检测器检测(参考条件:漂移管温度105℃,载气流速为2.6L/min,可根据具体情况适当调整条件)。精密量取对照溶液(1)、(2)和(3)各10μL,分别注入液相色谱仪,记录色谱图,以对照溶液浓度的对数值与相应的峰面积的对数值计算线性回归方程,相关系数(r)应不小于0.99;另取供试品溶液,同法测定,记录色谱图至主成分峰保留时间的2倍,供试品溶液色谱图中如有杂质峰,扣除硫酸峰,用线性回归方程计算供试品中庆大霉素C1a的含量。具体色谱参数参照依据中国药典2015年版二部硫酸依替米星项下有关物质第二法(LC-ELSD法)[7]。

表1 本实验所用材料Tab.1 Materials used in this experiment

表2 本实验所用仪器Tab.2 Instruments used in this experiment
(2)庆大霉素C1a的纯度测定:精密称取本品适量,用水定量稀释制成约含庆大霉素C1a 2.0mg/mL的溶液,作为供试品溶液;另取庆大霉素C1a对照品适量,精密称定,加水溶解并定量稀释制成约含庆大霉素C1a 2.0mg/mL的溶液,作为对照溶液,西索米星和小诺霉素的对照溶液配制方法相同;精密量取对照溶液适量,用水定量稀释制成含庆大霉素C1a各20、50和100μg/mL的溶液作为对照溶液(1)、(2)和(3)。HPLC-ELSD方法同“(1)”,精密量取对照溶液(1)、(2)和(3)各20μL,分别注入液相色谱仪,记录色谱图,以对照溶液浓度的对数值与相应的峰面积的对数值计算线性回归方程,相关系数(r)应不小于0.99;另取供试品溶液,同法测定,记录色谱图至主成分峰保留时间的2倍,供试品溶液色谱图中如有杂质峰,扣除硫酸峰,用线性回归方程计算供试品中庆大霉素C1a纯度。供试品溶液色谱图中小于对照溶液主峰面积0.02倍的峰忽略不计。
1.2.2 大孔吸附树脂柱层析
(1)色谱型大孔吸附树脂的初筛:选用HZ色谱填料-50、色谱3号、NM100、NM200色谱型大孔吸附树脂,每种树脂经预处理后分别装1根柱体积约300mL的高径比4:1的常压层析柱纯化庆大霉素C1a粗品。上样液pH调至11.5,上样量3g庆大霉素C1a粗品/ 100mL湿树脂,上样流速1BV/h,然后去离子水冲洗约3BV,流速0.2BV/h。接着采用pH1.6稀硫酸水解析,解析流速1.0BV/h,分步收集,每1BV收集1瓶,检测收集液的比旋度,有比旋度的收集液用HPLC检测含量和纯度。考察不同树脂纯化庆大霉素C1a粗品后获得纯度大于96%收集液样品的收率。
(2)NM200大孔吸附树脂柱层析参数的优化:①解析pH值的优化 各取NM200树脂500mL装入3根与“(1)”相同规格的色谱柱备用。分别取3份15g庆大霉素C1a粗品溶解于300mL蒸馏水中,调节pH11.5上柱吸附,流速1BV/h,然后去离子水冲洗约3BV,流速0.2BV/h。选取pH 1.8、2.0、2.2的稀硫酸水溶液解析,解析流速1.0BV/h,考察最优解吸条件。洗脱液分步收集,每1BV收集1瓶,检测收集液的比旋度,有比旋度的收集液用HPLC检测含量和纯度。考察不同解析pH下纯化庆大霉素C1a粗品后获得纯度大于96%收集液样品的收率。
②解吸流速的优化:各取NM200树脂500mL装入3根与“(1)”相同规格的色谱柱备用。分别取3份15g庆大霉素C1a粗品溶解于300mL蒸馏水中,调节pH11.5上柱吸附,流速1BV/h,然后去离子水冲洗约3BV,流速0.2BV/h。选取“①”条件较优的稀硫酸水溶液解析,考察不同解析流速0.5、1.5和2.0BV/h对树脂纯化效果的影响。洗脱液分步收集,每1BV收集1瓶,检测收集液的比旋度,有比旋度的收集液用HPLC检测含量和纯度。考察不同解析流速下纯化庆大霉素C1a粗品后获得纯度大于96%收集液样品的收率。
1.2.3 盐析结晶法精制庆大霉素C1a
(1)有机溶剂的种类和比例的初筛:分别选取甲醇、乙醇、丙酮、异丙醇等与水互溶的有机溶液,与10mL含庆大霉素C1a粗品300mg/mL的水溶液以4:1、7:1、10:1的比例混合均匀,加入浓硫酸调节pH至6.0~7.0后于室温中磁力搅拌3h考察庆大霉素C1a结晶情况。如有固体析出,送HPLC检测其纯度。
(2)加入硫酸调节pH先后顺序的考察:选取2份10mL含庆大霉素C1a粗品300mg/mL的水溶液,按照“(1)”结晶较好的溶剂种类和7:1比例条件,一份与溶剂混合后加入浓硫酸调节pH至6.0~7.0,室温中磁力搅拌3h考察庆大霉素C1a结晶情况;一份加入浓硫酸调节pH至6.0~7.0后与溶剂混合,室温中磁力搅拌3h考察庆大霉素C1a结晶情况。如有固体析出,送HPLC检测其纯度。
(3)有机溶剂混合比例的优化:选用“(1)”较优的两种溶剂,按照甲醇:乙醇 1:1、1:2、2:1、1:4、4:1的比例混合,分别与10mL含庆大霉素C1a粗品300mg/mL的水溶液以7:1的比例混合均匀,加入浓硫酸调节pH至6.0~7.0后于室温中磁力搅拌3h考察庆大霉素C1a结晶情况。如有固体析出,送HPLC检测其纯度和收率。
(4)结晶温度及结晶体系pH值的优化:按照“(3)”中较优溶剂条件分别与5组10mL含庆大霉素C1a粗品300mg/mL的水溶液混合后调节pH至6.0~7.0分别在30℃、室温、10℃、0℃、-10℃下磁力搅拌3h考察庆大霉素C1a结晶情况。如有固体析出,送HPLC检测其纯度。
按照“(3)”中较优溶剂条件分别与3组10mL含庆大霉素C1a粗品300mg/mL的水溶液混合后室温下,按照其硫酸与庆大霉素C1a的摩尔的量比为2:1、3:1、4:1加入混合液,磁力搅拌3h考察庆大霉素C1a结晶情况。如有固体析出,送HPLC检测其纯度。
(5)考察不同酸对其结晶形态及纯度影响:按照“(4)”中较优的结晶条件,分别与3组含庆大霉素C1a粗品300mg/mL的水溶液按照7:1混合室温加酸结晶,分别考察甲酸、乙酸、磷酸、盐酸和碳酸(通CO2气体),按照物质的量摩尔比2:1的比例加入即分别加0.56、0.50、0.58和0.75mL到混合溶液中,磁力搅拌3h考察庆大霉素C1a结晶情况。如有固体析出,送HPLC检测其纯度。其中结晶状态较好的检测其电子扫描显微镜形态和X射线粉末晶体衍射。
(6)电子扫描显微镜形态观察和X射线粉末晶体衍射检测方法:电子扫描显微镜形态观察条件为:使用导电胶,将样品粉末均匀黏附在铜台上。除去多余粉末,对样品溅射喷金30s后,用扫描电镜进行观察。X射线粉末晶体衍射的测定条件为:CuKa 40kV 40mA为光源,步长0.02°,扫描速度:8°/min,扫描范围:3°~80°,室温。
1.2.4 大孔吸附树脂和盐析结晶工艺组合优化
由于最终需要获得庆大霉素C1a游离碱形态,而上述纯化过程均会引入无机盐,因此在上述精制过程后需要一步离子交换树脂柱层析除盐过程;最终通过减压真空浓缩除氨水,并冷冻干燥即可获得高纯度庆大霉素C1a游离碱冻干粉。
所以最终的工艺路线组合可以有路线A和路线B,详见图1,分别进行上述实验后比较两种路线纯化后获得庆大霉素C1a游离碱冻干粉的纯度及纯化过程的总收率。
2 结果与分析
2.1 大孔吸附树脂柱层析
2.1.1 色谱型大孔吸附树脂的初筛(图2)
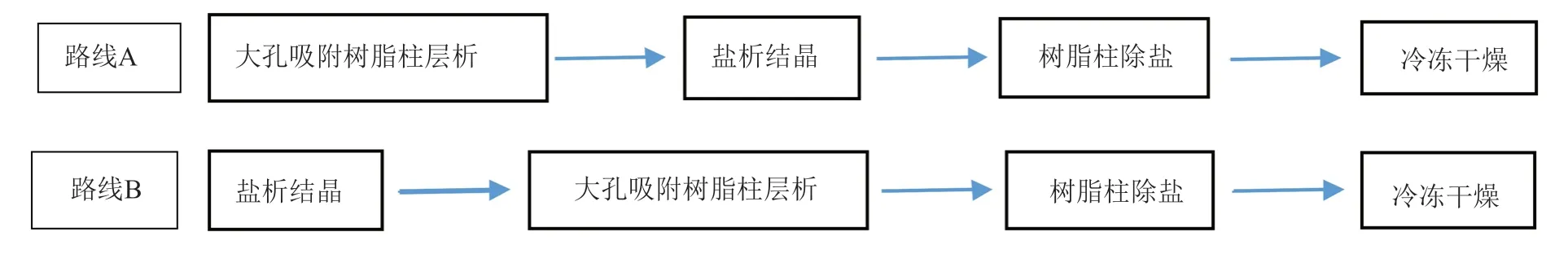
图1 组合优化的两种工艺路线Fig.1 Combinatorial optimization of two routes
根据前期依替米星纯化经验[8],4种色谱型大孔吸附树脂HZ-色谱填料50、色谱3号、NM100和NM200均能吸附庆大霉素C1a游离碱,水洗去除水溶强极性杂质后,pH1.6稀硫酸洗脱后,能去除主要杂质西索米星和小诺霉素。4种树脂纯化后HPLC纯度96%以上庆大霉素C1a的收率只有50%~60%,损失较大;其中NM200树脂纯化效果最佳。
2.1.2 NM200大孔吸附树脂柱层析参数的优化

图2 色谱型大孔吸附树脂初筛Fig.2 Chromatographic macroporous adsorption resin screening
(1)解析pH值的优化(图3):选取“2.1.1”项中最优树脂NM200进行解析pH的优化,pH1.6条件下解析较快,分离度不佳,造成HPLC纯度96%以上庆大霉素C1a的收率都低于60%。适当提高pH后,pH2.0和pH 2.2解析条件下,收率大于70%。但pH2.2解析时间较长,且与pH2.0相比,无显著差异。因此,选取pH 2.0作为较优的解析条件。
(2)解吸流速的优化:选取“(1)”最优解析pH2.0进行解析流速的考察,其中流速1.0和1.5BV/h的收率差异不大;最佳流速为0.5BV/h,HPLC纯度96%以上庆大霉素C1a的收率达到76%。由图4可知,最优解析流速为0.5BV/h。
2.2 盐析结晶法精制庆大霉素C1a
2.2.1 有机溶剂的种类和比例的初筛(表3)

图3 NM200树脂解析pH值优化Fig.3 Optimization of NM200 resin elution pH value
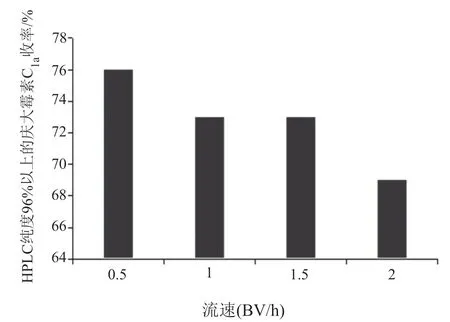
图4 NM200树脂解析流速的优化Fig.4 Optimization of NM200 resin elution flow rate

表3 有机溶剂的种类及与水溶液比例初筛Tab.3 Type and mixing ratio of organic solvent with aqueous solution
由于庆大霉素C1a水溶性极强,因此选取四种强极性有机溶剂甲醇、乙醇、丙酮和异丙醇进行盐析结晶不良溶剂的筛选。如表3所示,只有甲醇和乙醇与含庆大霉素C1a粗品300mg/mL的水溶液7:1和10:1互溶时能产生白色固体,其余条件都是油状物。将上述获得的白色固体过滤烘干后送HPLC检测庆大霉素C1a纯度分别为97.1%、97.3%、97.3%和97.0%,说明盐析结晶过程能大幅度提高庆大霉素C1a纯度。
2.2.2 加入硫酸调节pH先后顺序的考察
由于庆大霉素C1a不同pH值条件下极性变化很大,因此pH值调节顺序是影响结晶状态的重要因素之一。通过比较加入硫酸调节pH值的顺序差异发现,预先调pH后加7:1甲醇时,产生的白色固体形成大的絮状物,析出状态较差;而先加7:1甲醇后调pH值时,产生的白色固体形成分散小颗粒状固体,析出状态较好。将上述获得的白色固体过滤烘干后送HPLC检测庆大霉素C1a纯度分别为97.1%和97.5%。从析出状态和析出固体纯度指标看,先加入不良溶剂后加入硫酸调pH值的条件更佳。
2.2.3 有机溶剂混合比例的优化
根据其他氨糖类盐析结晶的经验[8-9],多种不良溶剂的混合结晶比例会对结晶纯度和收率产生一定的影响。分别按照两种不良溶剂甲醇与乙醇1:1、1:2、2:1、1:4、4:1的比例混合,并以7:1比例与含庆大霉素C1a粗品300mg/mL的水溶液混合盐析结晶,结果如表4所示。其中甲醇与乙醇1:2条件与单一溶剂甲醇和乙醇相比,庆大霉素C1a提高了0.8%左右;纯化收率都在93%以上。
2.2.4 结晶温度及结晶体系pH值的优化
考察结晶温度时,在30℃、室温、10℃时其有结晶状态较好,获得固体的庆大霉素C1a纯度分别为97.9%、98.1%和97.5%。而在0℃及-10℃时,其结晶状态较差,获得固体的庆大霉素C1a纯度分别为97.7%、97.3%。结果显示,室温时结晶状态较好,获得的庆大霉素C1a纯度最高。
考察结晶体系pH值时,按照硫酸和庆大霉素C1a摩尔比2:1(pH6.5左右)、3:1、4:1增加硫酸添加量后,获得的庆大霉素C1a纯度依次为98.0%,97.7%,97.8%。结果说明硫酸添加量对结晶纯度影响较小。
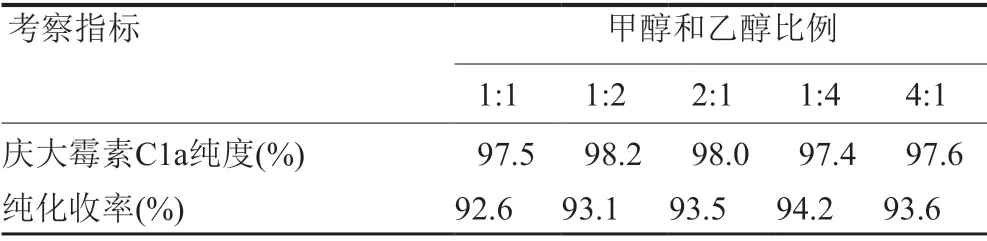
表4 甲醇和乙醇混合比例对析出庆大霉素C1a纯度和收率的影响Tab.4 Effect of mixing ratio of methanol and ethanol on the purity and yield of Gentamicin C1a
2.2.5 考察不同酸对其结晶形态及纯度影响(图5)
除硫酸外,其余几种无机酸也可能与庆大霉素C1a盐析产生较好的结晶物。考察了甲酸、乙酸、盐酸、磷酸和碳酸(通CO2气体),其中甲酸、乙酸和盐酸不析出固体,无法产生晶体。而磷酸和碳酸及上述主要研究的硫酸都能析出白色固体,但PXRD和电镜照片显示其结晶有序性较差,不是多晶型状态。磷酸和碳酸析晶固体的庆大霉素C1a纯度分别为95.2%和96.6%。

图5 磷酸、碳酸和硫酸盐析固体扫描电镜照品和PXRD图谱Fig.5 Phosphoric acid,carbonic acid and sulfate gentamicin C1a solid scanning electron microscopy and PXRD map
2.3 大孔吸附树脂和盐析结晶工艺组合优化
按照路线A和路线B进行两种路线组合优化(表5),比较大孔吸附树脂柱层析和盐析结晶工序顺序对最终产品纯度和纯化收率的影响。
结果显示(图6),路线B 获得的样品纯度为99.5%,收率大于70%;具有很强的产业化潜力。主要成本在树脂工序的高纯度样品损失和树脂的一次性投入。
3 结论
为了制备高纯度庆大霉素C1a游离碱,本论文摸索了大孔吸附树脂法和盐析溶媒析晶法来进一步纯化HPLC纯度为91%~93%的庆大霉素C1a游离碱粗品。
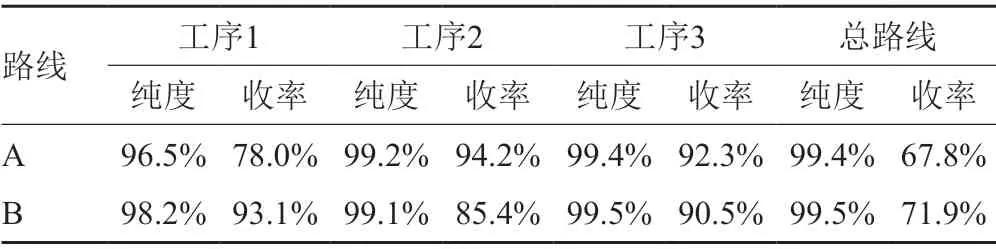
表5 路线A和路线B每个工序样品纯度和纯化收率Tab.5 Route A and route B purity and purification yield of each process sample
(1)通过筛选色谱型大孔吸附树脂,获得较优的树脂NM200;优化了影响层析收率的主要影响因素解析液pH值和解析流速,获得较优的解析液pH值为2.0和流速0.5BV/h。经过优化后,纯化收率从65%提高至74%。
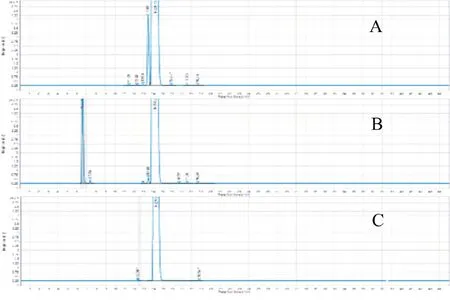
图6 庆大霉素C1a样品的HPLC-ELSD纯度检测图谱Fig.6 HPLC-ELSD purity detection map of gentamicin C1a sample
(2)通过筛选有机溶剂的种类和比例,确定较优的不良溶剂为甲醇和乙醇;比较硫酸添加顺序、甲醇和乙醇混合比例,确定先加溶剂后加硫酸和甲醇与乙醇1:2的条件较优,优化后获得的庆大霉素C1a纯度达到98.2%,收率大于93%。进一步优化结晶温度和结晶体系pH值,发现室温条件下硫酸和庆大霉素C1a摩尔比2:1(pH6.5左右)条件较优。
(3)研究了多种无机酸盐析结晶的情况,发现磷酸、硫酸和碳酸条件下能析出白色固体。但电子扫描电镜和PXRD结果显示,结晶状态较差,未达到多晶型状态。
(4)通过比较大孔吸附树脂和盐析结晶的纯化顺序变化,组合纯化后获得的庆大霉素C1a游离碱纯度大于99.0%,比纯化前样品提高6%以上,收率大于70%。
