IgG4相关性皮肤病三例报道并文献复习
2019-10-22贾倩楠刘桂丽
贾倩楠 刘桂丽 贾 力 方 凯 彭 军 渠 涛
IgG4相关性疾病(IgG4-related disease, IgG4-RD)由Kamisawa等[1]于2003年提出,是一类新近被认识的疾病,多器官受累,主要累及胰腺、唾液腺、泪腺、淋巴结和肺脏等部位。由于这类疾病比较少见且无特异性临床表现,长期误诊常常造成重要脏器功能损害,因此尽早诊断至关重要。2013年Sato等[2]首先报道这类疾病可以累及皮肤。Ingen-Housz-Oro等[3]同年报道两例仅有皮损的IgG4-RD。由于皮疹易于发现且可能是IgG4-RD的首发症状,因此对于IgG4相关性皮肤病的警惕具有重要意义。本文报道3例确诊皮肤受累的病例,并进行文献复习。
1 临床资料
病例1,女,65岁。因“双耳后瘙痒性肿块3年,伴双上眼睑肿胀”于2017年3月7日就诊。患者3年前无诱因右耳后出现结节,缓慢增大,左耳后逐渐出现类似皮疹。双侧眼睑间断性肿胀。外院拟诊“木村病”。因浅表淋巴结肿大,行右腋窝淋巴结活检,未予明确诊断。患者无发热、体重减轻等全身症状。
皮肤科查体:左耳后肿物,约3 cm×2.5 cm,质韧,表面发红;右耳后红色结节,约1.5 cm×1.2 cm,质硬,表面光滑;双上眼睑肿胀,轻度发红(图1a、1b)。颈部可触及多发轻度增大淋巴结。
实验室检查:血常规WBC 5.66×109/L[(3.5~9.5)×109/L],Eos 0.54×109/L[(0.5~5.0)×109/L],血红蛋白及血小板正常范围;ESR、血清IL-6及IL-10正常范围;免疫固定电泳未见M蛋白;IgG 22.24 g/L(7~17 g/L),IgM和IgA 正常范围,IgE 1775.0 KU/L(0~60 KU/L);IgG亚类:IgG1和IgG3正常范围,IgG2 7190 mg/L(1500~6400 mg/L),IgG4 14000 mg/L(80~1400 mg/L);β2微球蛋白2.16 mg/L(0.7~1.8 mg/L);感染指标:CMV-IgM(+);CMV-DNA 2000copies/mL;EBV IgA/EA(+)。
唾液腺B超示双侧颌下腺回声不均,双侧腮腺内及双侧颌下多发淋巴结肿大伴回声异常。胸增强CT示右侧腋窝多发肿大淋巴结,两肺门及纵隔多发小淋巴结,右肺上叶、左肺上叶以及双肺下叶多发淡片及索条影。腹盆腔增强CT示胆总管扩张,腹膜后、盆腔及双侧腹股沟未见增大淋巴结影。
皮肤组织病理示:表皮无特殊改变,真皮内淋巴细胞团块状浸润,可见少量嗜酸粒细胞及浆细胞(图1c、1d)。免疫组织化学染色示CD138部分细胞(+),IgG4阳性细胞平均80个/高倍视野(图1e)。淋巴结活检:淋巴结结构存在,皮质区内淋巴滤泡增生,可见散在浆细胞。IgG4/IgG大于40%。
治疗:醋酸泼尼松龙片20 mg日1次,雷公藤多苷片20 mg日2次,阿法骨化醇软胶囊0.25 μg日1次。1年后皮损减轻,醋酸泼尼松龙片剂量调整为10 mg 日1次。19个月随诊时病情反复,总IgE 646.0 KU/L,IgG2 6580 mg/L,IgG4 5660 mg/L。
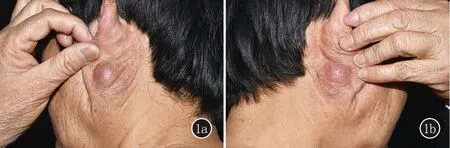
图1 1a,1b:病例1左耳后及右耳后红色浸润性结节;1c、1d:病例1真皮内淋巴细胞团块状浸润,可见嗜酸粒细胞及浆细胞 (HE,×40;×200);1e:IgG4部分细胞阳性(SP,×200)

病例2,男,51岁。因“面、颈及前胸瘙痒性浸润性红斑块及结节4年”于2015年12月21日就诊,否认口干、消化道症状及系统性症状。外院查血常规显示嗜酸粒细胞升高,经治疗(具体药物不详)嗜酸粒细胞恢复正常范围。
皮肤科查体:右侧面颊、颈部及前胸浸润性红斑块及结节,有融合倾向,双上肢伸侧散在浸润性丘疹(图2a)。淋巴结未触及明显异常。
实验室检查:免疫固定电泳未见M蛋白;免疫球蛋白IgE 3495.0 KU/L,IgG、IgA和IgM正常,IgG亚类IgG1和IgG2正常,IgG3(127 mg/L)降低,IgG4(5170 mg/L)升高;抗核抗体阴性;血清IL-6和IL-8正常范围。
皮肤组织病理示:真皮内血管周围淋巴细胞、组织细胞、浆细胞及嗜酸粒细胞灶状浸润(图2b、2c)。免疫组织化学染色示IgG4阳性细胞平均45个/高倍视野(图2d)。
治疗:甲泼尼龙8 mg日2次,雷公藤多苷片20 mg日3次,沙利度胺片25 mg日3次,烟酰胺片50 mg日3次,外用吡美莫司乳膏,5个月随访时症状减轻,皮疹变薄,9个月随访,皮损减轻;实验室检查示IgE 1064.0 KU/L,IgG4 531 mg/L,均有显著下降。
病例3,女,61岁。因“面部及前胸瘙痒性结节及斑块1年”于2019年6月5日就诊。患者颈部淋巴结肿大伴口眼干燥10年,外院血常规显示嗜酸粒细胞升高,淋巴结活检显示淋巴组织反应性增生,建议排除IgG4相关性疾病,并于4年前给予泼尼松40 mg日1次及阿法骨化醇软胶囊0.25 μg日1次,症状好转,但停药后复发。
皮肤科查体:右侧眼部周围、面颊及下颌部位散在暗红色质韧丘疹、结节,左下颌角处少量红色结节,前胸局部可见类似皮损(图3a)。
实验室检查:血常规正常范围;ESR 62 mm/h (0~20 mm/h);免疫固定电泳未见M蛋白;IgG、IgM和IgA正常范围;IgE 277.0 KU/L;IgG亚类:IgG1~IgG3正常范围,IgG4 17100 mg/L;IL-6正常范围;ANA(IgG型)散点型1∶320,核膜型1∶160;抗着丝点抗体(IgG型)1∶320;抗可溶性核抗原(ENA)抗体均阴性;其余自身抗体均阴性。
皮肤组织病理示:真皮血管周围淋巴细胞及浆细胞结节状分布,血管壁增厚,成纤维细胞同心圆状增生,血管腔变窄,病变累及皮下脂肪血管周围(图3b、3c)。免疫组织化学染色示IgG4阳性细胞平均81个/高倍视野(图3d)。
口咽部核磁显示两侧腮腺信号不均匀,其内多发卵圆形异常信号,高度怀疑肿大淋巴结,右侧颌下区及两侧颈部血管旁多发肿大淋巴结。胰腺核磁扫描未见异常。
治疗:甲泼尼龙20 mg日1次,骨化三醇软胶囊0.25 μg日1次,外用硼锌糊。
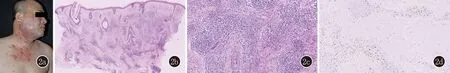
图22a:病例2右侧面颊、颈部及前胸红色浸润性斑块及结节;2b、2c:病例2真皮内血管周围淋巴细胞、组织细胞、浆细胞及嗜酸粒细胞灶状浸润(HE,×100;×200);2d:IgG4阳性(SP,×200)
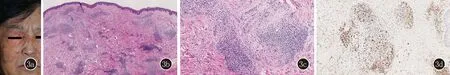
图33a:病例3右侧眼部周围、面颊及下颌部位散在暗红色质韧丘疹结节;3b、3c:病例3真皮血管周围淋巴细胞及浆细胞结节状分布,血管周围成纤维细胞同心圆状增生(HE,×100;×200);3d:IgG4阳性(SP,×200)
2 讨论
IgG4-RD是一种系统性疾病,主要累及中老年人,以合成分泌IgG4的浆细胞浸润以及血清高水平IgG4为主要特征[4],引起自身免疫性疾病,造成受累器官或组织的肿胀、硬化及功能障碍。IgG4-RD的发病与遗传、微生物、自身抗体、固有免疫和适应性免疫等多因素有关,但具体机制仍不清楚。IgG4-RD最常累及的器官是胰腺,引起自身免疫性胰腺炎,该病是1991年由Kawaguchi等[5]首次报道的,主要临床表现为梗阻性无痛性黄疸,为自身免疫引起的胰腺慢性炎症性病变。此外还可累及泪腺、唾液腺、肺脏、淋巴结、肾脏、肝脏、胆管、脑膜、腹膜后、乳腺、垂体、甲状腺、前列腺和皮肤[4]。
IgG4-RD的组织学特征包括淋巴细胞及浆细胞浸润,席纹状纤维组织增生,嗜酸粒细胞浸润以及闭塞性血管炎[4]。本病的正确诊断需要结合病史、体格检查、实验室检查、影像学检查以及最重要的组织病理学检查。2011年Umehara等[6]提出IgG4-RD的诊断指标包括:临床上一个或多个器官出现病变;外周血IgG4>1350 mg/L;组织中IgG4+细胞>10个/高倍视野,IgG4+/IgG+浆细胞比值>40%。
IgG4-RD可以累及单个器官,也可以多器官受累,然而IgG4-RD的皮肤受累较少见。在有些病例中,皮肤受累是本病多系统损害的皮肤表现;而有些情况下,皮肤受累是IgG4-RD的首发症状,但是常常在其他器官受累后才得以确诊。因此,对于IgG4+细胞和血清水平升高的皮肤浸润型患者,应考虑到IgG4-RD的可能。目前,IgG4-RD出现皮肤损害的具体机制还不明确,可能是由于浆细胞浸润或IgG4介导的炎症所致。
本文三例患者临床表现为瘙痒浸润性结节或斑块,分布于面颊、眼睑、耳周、颈部及胸部,这些临床表现与Sato等报道的经典表现一致[2]。但是,Tokura等[4]于2014年将IgG4相关性皮肤病分为两大类,一类是由于IgG4+浆细胞浸润所致,如皮肤浆细胞病、假性淋巴瘤及淋巴血管样增生伴嗜酸粒细胞增多、米枯力兹病;另一类是IgG4介导的炎症反应所致,如银屑病样皮疹、非特异性斑丘疹和红斑、高免疫球蛋白血症性紫癜和荨麻疹样血管炎、缺血性肢端。这些病变多数为描述性诊断,与本组的临床表现不完全类似,因此,需要更多的研究和随诊来确立IgG4相关性皮肤病的诊断标准。
IgG4-RD的组织病理表现被分为两种组织学类型:结节性皮炎型和皮下结节型。细胞浸润的位置不同,分别为真皮或皮下,但浸润的细胞类型相似,都有丰富的浆细胞、小淋巴细胞和嗜酸粒细胞,其中浆细胞以IgG4+浆细胞为主,有研究发现IgG4+浆细胞计数为49~396个/高倍视野(平均值±SD,172±129),IgG4+/IgG+细胞比为62%~92%[2]。
IgG4被普遍认为是非致病的温和性抗体,与 C1q和Fc受体的亲和力低,不能通过经典途径激活补体,不能引起严重的免疫反应;而且IgG4只能与抗原形成单价键,难以形成较大的免疫复合物,这些因素均降低了引发免疫损伤的可能性[7]。然而,研究发现在天疱疮和大疱性类天疱疮中,IgG4是其主要的抗体成分,沿表皮真皮连接处分布,与致病性密切相关[8]。在天疱疮中,IgG4抗体可通过与桥粒中的靶抗原结合而引起表皮内水疱,而不依赖补体和白细胞的激活[9]。从地方型落叶性天疱疮患者体内纯化的IgG4抗体,已被证明对小鼠具有致病性[10]。同样,大疱性类天疱疮等表皮下疱病的IgG4在离体皮肤模型中,可诱导真表皮分离[11]。研究发现,IgG4-RD患者血清 IgG4水平随激素的升高而波动,与疾病严重程度不呈平行关系,也无明显相关性,提示 IgG4并不直接参与疾病症状的发生,也不能作为预测疾病活动度的可靠指标[12]。但是,本组病例1和病例2经数月治疗后,血清IgG4水平明显下降。因此我们认为,天疱疮或大疱性类天疱疮作为仅累及皮肤的自身免疫性疾病,IgG4在其发病中起重要作用,但IgG4-RD作为系统性疾病,研究IgG4的致病作用将会更加复杂,仍需进一步的研究工作。
本组三个病例均显示外周血嗜酸粒细胞和IgE升高,组织病理中出现嗜酸粒细胞及血管周围纤维化。研究发现,IgG4-RD患者体内通常有高水平的T细胞分泌的细胞因子,如IL-4、IL-5、IL-10和TGF-β。Th2细胞通过分泌IL-4、IL-5和IL-13促进活化的B细胞生成IgG4和IgE,促进IgG向IgE的类别转换,同时促进嗜酸粒细胞的生长、活化和分化[4]。实际上,40%的IgG4-RD患者的血清学检查显示高水平的IgE和嗜酸粒细胞[2]。另外Th2和Tr1细胞分泌的IL-10,以及Th3细胞分泌的TGF-β,能促进活化的B细胞合成IgG4,参与受累器官中IgG4+浆细胞的浸润[13]。同时TGF-β已被广泛认为能够诱导纤维化,可能是引起IgG4-RD组织纤维化改变的重要原因[3]。
有文献报道,单器官受累的IgG4-RD患者血清IgG4水平可能在正常范围内,推测高水平的IgG4可能只是系统性IgG4-RD的标志物[6]。还有研究发现,仅仅出现IgG4+浆细胞和/或血清中IgG4水平升高,可发生在其他非IgG4相关性疾病中,包括一些不同类型的皮炎[14]。因此,鉴别诊断在IgG4-RD的诊断中起重要作用,IgG4-RD皮肤受累的主要鉴别诊断包括淋巴组织增生性疾病(如皮肤假性淋巴瘤、B细胞淋巴瘤,浆细胞型多中心Castleman病及Rosai-Dorfman病),慢性纤维性血管炎(如面部肉芽肿和持久性隆起性红斑)等。
假性淋巴瘤在临床和组织病理上均与IgG4相关性皮肤病相似。假性淋巴瘤皮疹同样好发于面部、前胸或上肢,表现为单个、群集或多发的红棕色或紫红色斑块或结节;组织学示真皮内淋巴细胞结节状或弥漫性浸润,伴有不同程度的组织细胞、嗜酸粒细胞和浆细胞浸润。然而,假性淋巴瘤常常出现“头重脚轻”的浸润模式,并可有含混合淋巴细胞的淋巴滤泡的形成,少见IgG4+浆细胞,可与IgG4相关性皮肤病鉴别。
多中心Castleman病是一种罕见的淋巴增殖性疾病,有报道浆细胞型多中心Castleman病患者血清中有高水平IgG4,甚至满足IgG4-RD的组织学诊断标准[15]。不过浆细胞型多中心Castleman病是一种高IL-6综合征,同时伴有贫血、低白蛋白血症、多克隆γ-球蛋白血症、血沉和CRP升高。高IL-6综合征仅凭血清学、临床表现及组织病理有时难与IgG4-RD鉴别,需要结合其他辅助检查,特别是实验室检查综合考虑。
面部肉芽肿和持久性隆起性红斑在组织病理上同样出现席纹状纤维化,浆细胞及嗜酸粒细胞浸润,免疫组织化学染色虽然显示IgG4+浆细胞,但不符合IgG4-RD的诊断标准。学者们基本认为这两种疾病不属于IgG4-RD,两者的合并疾病和预后均与IgG4-RD有明显差异[16]。
Rosai-Dorfman病,又称伴巨大淋巴结病的窦组织细胞增生症,以淋巴结内窦组织细胞浸润为主要的病理特点。皮肤Rosai-Dorfman病是一种特殊类型,通常表现为良性病程,部分皮损可自发消退。临床表现多种多样,表现为红色或黄瘤样丘疹、结节或斑块。组织病理表现为真皮和浅层皮下组织组织细胞、淋巴细胞、中性粒细胞、浆细胞及嗜酸粒细胞弥漫浸润,可以出现纤维组织增生。炎症细胞位于组织细胞浆内,称为伸入运动,是本病特征性的病理表现。尽管皮肤Rosai-Dorfman组织病理具有IgG4+浆细胞和真皮纤维化的特点,但仍不符合IgG4-RD的诊断标准[17]。
IgG4-RD是新近认识的一类系统性疾病,尚无统一的治疗方案。目前,最有效的治疗方案多为系统性应用糖皮质激素,但在糖皮质激素停用或者减量后可能复发,需要定期监测[18]。文献报道的非激素治疗方案主要包括沙利度胺,雷公藤多苷片,以及生物制剂,如利妥昔单抗和英夫利昔单抗。同时,患者需要长期密切随诊,及时发现其他器官的受累。
