使用R语言metaMA程序包实现基因表达谱数据的Meta分析
2019-09-27李贤菁邓巧玲李嘉兴张晨曦阙雅婷翁鸿黄静宇李胜
李贤菁,邓巧玲,李嘉兴,张晨曦,阙雅婷,翁鸿,黄静宇,李胜,3,4
基因芯片技术又称微阵列技术[1],因其具有操作简单,信息量大,响应迅速等特点,在医学和生物学等领域得到广泛运用。DNA芯片属于基因芯片的一种类型,它是将已知序列的核苷酸作为探针,高密度排布在固相载体上。荧光标记的cDNA与载体上的已知序列杂交,用相应计算机软件检测和处理荧光信号强度,从而获得基因特征性序列或者特异性表达的相关生物信息。
随着分子生物学和基因组学的深入研究,基因芯片技术不断得到发展和完善。芯片尺寸增大,种类与数据类型也愈加丰富。而如何能够有效的处理和分析来自基因芯片的大量数据,也是研究者们关注的焦点。R是一款功能强大的统计分析软件,拥有完整的数据处理、统计和绘图系统以及操作环境。在生物信息方面,研究者通过开发不同R包,以满足分析不同类型基因芯片数据的需要。
基于R语言的metaMA包由Guillemette Marot开发[2]。不同研究中通过比较正常组织和癌组织不同基因表达量的差异而筛选出的差异表达基因不完全相同,使用metaMA包进行meta分析筛选出差异表达基因的方法基于更大样本量,因此可增加结论的统计功效。此文中涉及metaMA分析流程的R语言程序包主要有GEOquery[3]、org.Hs.eg.db[4]、metaMA[2]、VennDiagram[5]、db1[6]、db2[7]、db3[8]、annaffy[9]。其中,GEOquery用于基因组数据的获取,org.Hs.eg.db用于实现数据转换,metaMA用于找出差异表达的基因,VennDiagram用于绘制多组研究中差异基因的维恩图,db1、db2和db3用于对3例基因组数据中的差异基因进行注释,annaffy用于生成已识别探针的生物信息。本文将以3例口腔鳞状细胞癌数据集为例,说明如何对公开的不同研究数据进行统一转化,继而运用metaMA包找出差异表达的基因,绘制维恩图,并通过对差异基因注释得出结论。
1 数据准备
1.1 软件R的安装及基因组数据读入 从官方网站(https://www.r-project.org)下载最新版R软件R-3.4.2并进行安装。获取GEO数据,可以使用R语言的GEOquery包。安装、加载GEOquery包,以及下载相应的口腔鳞状细胞癌数据集。具体代码如下:
source("https://bioconductor.org/biocLite.R")
biocLite("GEOquery")
library(GEOquery)
data1<- getGEO('GSE9844')
data2 <- getGEO('GSE3524')
data3 <- getGEO('GSE13601')
数据集可以直接从GEO(https://www.ncbi.nlm.nih.gov/geo/)下载txt文件,用read.table()读入R,或者通过Excel整理出如表二所示的基因表达矩阵,用read.csv()读入R。
1.2 示例数据集 以3例口腔鳞状细胞癌数据集为例,说明metaMA的分析流程。3例数据集如表1所示。

表1 示例3例口腔鳞状细胞癌数据集
1.3 芯片预处理 基因芯片预处理过程,即是根据
荧光信号生成的数据将其转变成基因组数据的过程。质量控制和数据处理对基因芯片分析的有效性至关重要。通过质量控制,可以剔除不合格芯片,保留合格芯片做后续的数据处理分析。数据处理包含以下步骤,分别是背景校正,标准化,汇总。背景校正用于去除背景噪声的影响。标准化用于消除样品制备等非实验因素对结果的影响。最后经过汇总得到探针组水平的数据。目前较为流行的预处理方法有dChip,MAS5,RMA。前两种的背景校正方法均为mas,后一种的背景校正方法为rma。
1.4 根据基因组数据绘制箱线图 在使用metaMA程序包进行Meta分析之前,需要查看以上三个数据集基因表达水平的分布情况,这里绘制各组基因表达数据的箱线图,能够直观比较预处理效果具体代码如下:
par(mfrow=c(2,2))
boxplot(data.frame(exprs(data1[[1]])),outline=FALSE)
boxplot(data.frame(exprs(data2[[1]])),outline=FALSE)
boxplot(data.frame(exprs(data3[[1]])),outline=FALSE)
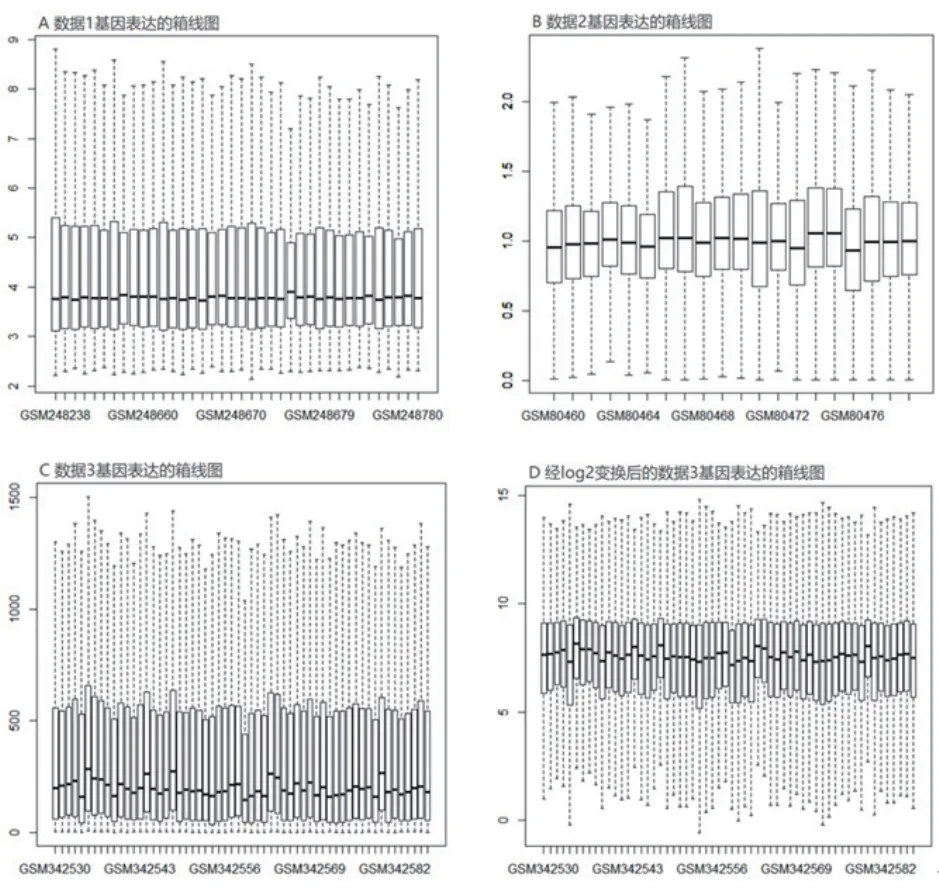
图1 箱线图
箱线图中,横坐标为样本,纵坐标为表达值,比较表达值从而获得结果变异程度的信息。从获得箱线图(图1A、1B和1C)中可看出,数据3(data3)的值远大于数据1(data1)和数据2(data2),变异相对较大。数据1[10]、数据2[11]与数据3[12]的数据处理方法不完全相同。数据1使用RMA方法对芯片数据做预处理。数据2使用Mas5.0软件对Affymetrix数据进行初步分析,进一步标准化数据后,输出到应用程序Genespring 6.0(Silicon Genetics,Redwood City,CA)。数据3使用Mas5.0方法的预处理,未进行对数转换进行标准化,这里我们对data3数据集进行2为底的对数转换,同时我们再次绘制标准化后的data3基因表达水平分布图(图1D),具体代码如下:
exprs(data3[[1]])<-log2(exprs(data3[[1]]))
boxplot(data.frame(exprs(data3[[1]])),outline=FALSE)1.5 明确样本分组 如前所述,本研究使用的3例口腔癌数据集包含正常组织和癌组织两类样本,在使用metaMA进行meta分析前,需要对以上两类样本进行分组、编码。我们将正常口腔组织编码为1,口腔癌组织编码为0。具体代码如下:
c1<-as.numeric(pData(data1[[1]])["source_name_ch1"]=="Oral Tongue Squamous Cell
Carcinoma")
c2<-as.numeric(apply(pData(data2[[1]])["descript ion"], 1,toupper)=="SERIES OF 16 TUMORS")
c3<-as.numeric(pData(data3[[1]])["source_name_ch1"]=="Tumor")
classes<-list(c1,c2,c3)
1.6 将基因表达矩阵转化为统一的Unigene形式如表1所示,这三个口腔癌基因表达谱分别采用不同平台的基因芯片进行检测,且三个数据集的探针集是不同的,因此需要将其转换成形式统一的基因编号。函数probe2unigene返回这3组独立研究中的探针所对应的所有UNIGENE。 函数unigene2probe将UNIGENE与Entrez Gene ID相对应。 要实现这一转换,需要加载Bioconductor中的org.Hs.eg.db包。具体代码如下:
source("http://bioconductor.org/biocLite.R")
biocLite("org.Hs.eg.db")
require("org.Hs.eg.db")
x <- org.Hs.egUNIGENE
mapped_genes <- mappedkeys(x)
link<- as.list(x[mapped_genes])
probe2unigene<-function(expset) {
#construction of the map probe->unigene
probes=rownames(exprs(expset))
gene_id=fData(expset)[probes,"ENTREZ_GENE_ID"]
unigene=link[gene_id]
names(unigene)<-probes
probe_unigene=unigene
}
unigene2probe<-function(map) {
suppressWarnings(x <- cbind(unlist(map),names(map)))
unigene_probe=split(x[,2], x[,1])
}
需要注意的是,无法与UNIGENE相匹配的探针将不被用于Meta分析。因为一个unigene可能会对应多个探针,选择一个恰当的方法来合并独立研究的探针至关重要。下面代码中,根据已被证实有效的参数mergemeth的默认值,convert2metaMA选择通过计算它们的平均值来总结对应于相同UNIGENE的探针的值。convert2metaMA函数为每个研究构建矩阵,矩阵中只包含所有研究里共有的UNIGENE对应的行(基于UNIGENE转换)。这些矩阵存储在函数返回的值的子集列表中。convert2metaMA还会返回conv.unigene,作用是在进行meta分析后能够返回原始探针的名称。具体代码如下:
convert2metaMA<-function (listStudies,mergeme th=mean) {
if (!(class(listStudies) %in% c("list"))) {
stop("listStudies must be a list") }
conv_unigene=lapply(listStudies,
FUN=function(x) unigene2probe(probe2unigene(x)))
id=lapply(conv_unigene,names)
inter=Reduce(intersect,id)
if(length(inter)<=0){stop("no common genes")}
print(paste(length(inter),"genes in common"))
esets=lapply(1:length(listStudies),FUN=function(i){
l=lapply(conv_unigene[[i]][inter],
FUN=function(x) exprs(listStudies[[i]])[x,,drop=T RUE])
esetsgr=t(sapply(l,FUN=function(ll) if(is.null(dim(ll))){ll}
else{apply(ll,2,mergemeth)}))
esetsgr
})
return(list(esets=esets,conv.unigene=conv_unigene))
}
conv<-convert2metaMA(list(data1[[1]],data2[[1]],data3[[1]]))
输出结果为
[1]"7745 genes in common"#说明此三个基因组中存在相交情况的基因数目位为7745。
esets<-conv$esets
conv_unigene<-conv$conv.unigene
上图中,表2为data1的基因表达矩阵部分,其中,第一行是样本编号,第一列为探针编号。表3为合并后的基因表达矩阵部分,第一列为探针对应的UNIGENE,数目即为三组研究中相交的基因数目。
2 应用metaMA包进行分析
加载metaMA包时需要同时用到包limma和包SMVar。包limma属于R的基础包之一。而包SMVar属于Bioconductor,如未安装,则需要安装之后才能顺利加载metaMA包。
source("http://bioconductor.org/biocLite.R")
biocLite("SMVar")
install.packages(“metaMA”)
library(metaMA)
res<-pvalcombination(esets=esets,classes=classes)
输出结果如表4
通过length函数,我们可以查看本次Meta分析所得出的差异基因的数目,具体代码如下:
length(res$Meta)
输出结果为
[1]2481#说明3组数据中,差异表达基因数目为2481。

表2 数据1的表达谱
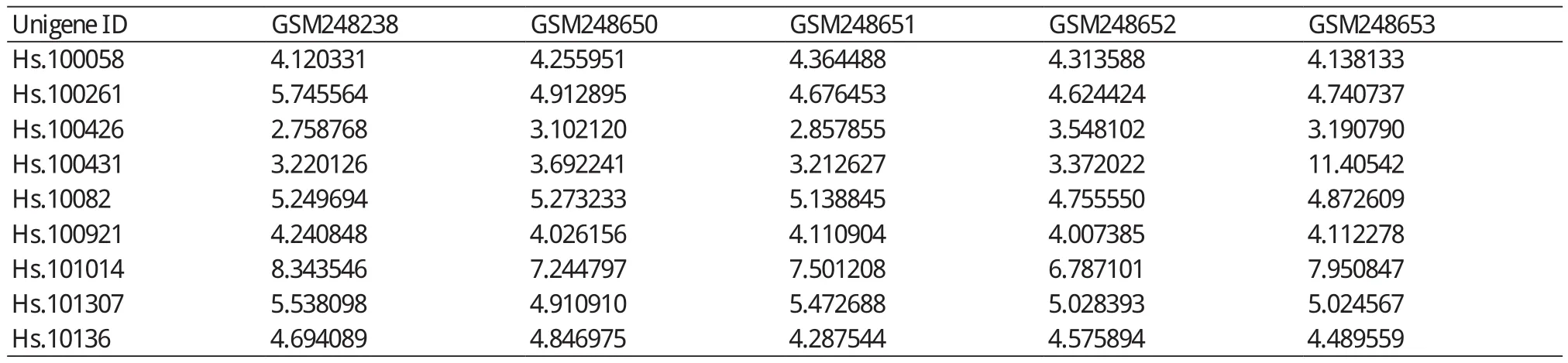
表3 转化为UNIGENE后的表达谱

表4 Meta分析后相关与差异基因相关的统计结果
此外我们还可以查看经Meta分析后的差异基因结果,具体代码如下:
Hs.Meta<-rownames(esets[[1]])[res$Meta]
3 绘制维恩图
通过绘制维恩图,可以直观比较独立研究或者Meta分析中差异表达基因的数目。安装VennDiagram包可以实现该功能。代码如下:
install.packages("VennDiagram")
library(VennDiagram)
venn.plot<-venn.diagram(x=list(study1=res$stud y1,study2=res$study2,
study3=res$study3, meta=res$Meta), filename =NULL, col = "black",
fill = c("blue", "red", "purple","green"),margin=0.05, alpha = 0.6)
jpeg("venn_jpeg.jpg")
grid.draw(venn.plot)
dev.off()
维恩图中,蓝色,紫色,红色区域分别表示在三个独立研究中筛选出的差异基因,绿色表示经Meta分析筛选出的2481个差异基因。由维恩图知,经由metaMA包筛选出而不能在独立研究中被识别的差异基因数目为219(图2)。
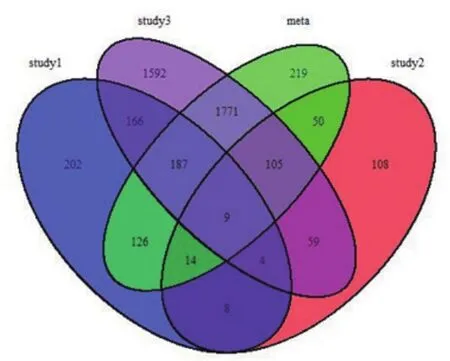
图2 经分析后的差异表达基因的维恩图
4 进一步筛选差异基因
经metaMA分析获得的2481个差异基因,需进一步进行筛选得出关键核心基因。可从TCGA数据库中下载头颈部腺癌的基因表达矩阵,根据临床信息从中筛选出口腔鳞状细胞癌数据纳入研究。本次研究共筛选出211个癌组织样本和27个正常组织对照。对其进行差异基因表达分析,获得P值<0.01的差异基因后,取这些基因与上述经metaMA分析获得的2481个基因的交集。将交集中的基因结合TCGA数据库提供的临床信息进行生存分析,以确定关键核心基因。
5 注释
5.1 获取芯片类型 在找到差异表达基因后,用getanndb函数获得相应的芯片类型。
getanndb<-function(expset) {
gpl_name=annotation(expset)
gpl=getGEO(gpl_name)
title=Meta(gpl)$title
db=paste(gsub("[-|_]","",
tolower(strsplit(title,"[[]|[]]")
[[1]][[2]])),"db",sep=".")
return(db)
}
db1<-getanndb(data1[[1]])
db1
#[1]"hgu133plus2.db"
db2<-getanndb(data2[[1]])
db2
#[1]"hgu133a.db"
db3<-getanndb(data3[[1]])
db3
#[1]"hgu95av2.db"
5.2 使用注释包对差异表达基因进行注释
#source("http://bioconductor.org/biocLite.R")
#biocLite(db1)
#biocLite(db2)
#biocLite(db3)
library(db1,character.only=TRUE)
library(db2,character.only=TRUE)
library(db3,character.only=TRUE)
5.3 获取注释文件 在下面代码中,origId.Meta可返回各独立研究中基因的原始IDs,原始IDs与进行Meta分析的共同差异表达基因的Hs.Meta IDs相对应。由于几个原始的探针匹配相同的UNIGENE,origId.Meta可能会比Hs.Meta包含更多的ID。annaffy包中的函数aafTableAnn和saveHTML可用于保存当前工作目录中的一个命名为“annotation.html”的 HTML文件,该文件包含本次分析的差异基因及其相应的生物信息。
origId.Meta<-lapply(conv_unigene,FUN=function(vec) as.vector(unlist(vec[Hs.Meta])))
source("http://bioconductor.org/biocLite.R")
biocLite("annaffy")
library(annaffy)
annlist<-lapply(1:length(origId.Meta),FUN=function(i)
aafTableAnn(origId.Meta[[i]],chip=get(paste0("db", i))))
annot<-do.call(rbind,annlist)
saveHTML(annot,file="annotation.html",title="Responder genes")
6 讨论
基因芯片技术发展迅速,被用于生物学、医学等多个领域研究。Meta分析是基于多项研究成果的荟萃分析。目前用于获取Meta分析所需的基因表达数据的主要平台包括GEO(https://www.ncbi.nlm.nih.gov/geo/)及ArrayExpress(http://www.ebi.ac.uk/arrayexpress/)等。按照常规研究思路,通过meta分析,筛选出与疾病相关的差异表达基因后,需要对差异表达的基因进行预测和功能分析。探究这些基因的潜在的生物学功能,有助于分析解释某些疾病的发生机理,为治疗提供新思路[13]。
严格的文章筛选和有效的质量评价是获得可靠结论的必要条件,此外还需要识别和控制发表偏倚。在输入Meta分析的研究数据时,需要将不同平台、不同类型芯片之间的差异纳入考虑范围。有时还需选择合适方法处理尚未进行预处理的数据。本文的3组基因组数据定量分析了口腔鳞状细胞癌相关基因的基因表达情况,并没有重点讨论标准化方法,仅是对数据3进行以2为底的对数转换。在进行严谨的基因芯片分析过程中,需要对数据处理方法进行评估和优化。
