儿童原发性皮肤骨瘤11例临床分析
2019-09-24徐教生向欣徐子刚邢嬛马琳
徐教生 向欣 徐子刚 邢嬛 马琳
首都医科大学附属北京儿童医院皮肤科100045
皮肤骨瘤(osteoma cutis)是指以真皮和/或皮下脂肪组织发生异位性骨化为特征的一组疾病,又称皮肤骨化[1]。皮肤骨瘤分为原发性和继发性,原发性皮肤骨瘤无前驱病变,约占总皮肤骨瘤的15%;继发性皮肤骨瘤在发病前存在其他病变,包括炎症(如硬皮病、皮肌炎或瘢痕等)、肿瘤(如黑素细胞痣、毛母质瘤等)、外伤或内分泌疾病等,约占皮肤骨瘤的85%[2]。临床上,皮肤骨瘤非常少见,而原发性皮肤骨瘤则更为少见,目前国内外文献以个案报道为主[3-5]。我们收集11例儿童原发性皮肤骨瘤,回顾其临床表现、组织病理及转归。
临床资料
一、一般资料
首都医科大学附属北京儿童医院皮肤科2011年8月至2018年8月确诊的原发性皮肤骨瘤11例,男7例,女4例;中位发病年龄1个月,3例出生时发病,7例于出生后1个月内发病,10例于出生后6个月内发病,1例发病年龄为22个月。4例早产,其中1例伴有宫内窘迫。所有患儿发病时均无发热,未见发育畸形,否认类似疾病家族史。
二、临床表现
皮损呈绿豆大小质硬丘疹、结节或融合成斑块,部分呈白垩色、质硬的粟粒样丘疹,皮损表面淡红或肤色,伴有轻度萎缩。3例皮损局限于1个部位,其中2例位于下肢,1例位于腹部,皮损为局限性融合性斑块,周围散在结节。8例皮损累及多个部位,其中7例累及躯干及四肢,2例累及头皮,未见面部受累;均有皮下质硬结节及斑块,2例伴有粟粒样丘疹,可自行破溃,1例右下肢、臀部大片融合性皮下质硬斑块,伴有患肢萎缩僵硬、屈曲受限,完全下蹲障碍。见图1、2。
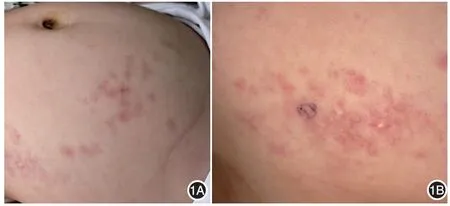
图1 1例原发性皮肤骨瘤临床表现

图2 1例进行性骨发育异常患儿临床表现及X线检查结果
三、实验室及影像学检查
所有患儿血清钙、甲状旁腺激素、抗核抗体及可溶性核抗原谱未见异常。9例血磷(1.84~2.03 mmol/L)轻微升高,3例碱性磷酸酶(255~259 U/L)及乳酸脱氢酶(299~362 U/L)轻度升高,4例肌酸激酶(203~231 U/L)轻度升高,6例天冬氨酸转氨酶(40.2~58.5 U/L)轻度升高,所有患儿丙氨酸转氨酶正常。11例均经内分泌科会诊排除原发性甲状旁腺功能减退或假性甲状旁腺功能减退。
患儿皮损部位X线检查:可见大小不等的高密度不规则钙化影,周围呈放射状,10例局限于皮肤层,1例可见皮下深层受累,形成新生骨。见图2。
四、组织病理检查及GNAS基因突变检测
11例患儿皮损组织病理均显示成熟板层骨形成,未见炎症细胞浸润,可见轻度表皮萎缩,骨化区域及周围组织内毛细血管增生及扩张显著。所有病例均累及真皮,5例侵及皮下脂肪组织,1例可见穿通现象。见图3。
3例患儿家属同意进行外周血GNAS基因检测,抽取3例患儿及其父母外周血后分别送至北京康旭医学研究所(2例,第1代测序方法)和北京迈基诺医学检验所(1例,全外显子测序)进行基因检测,结果显示3例患儿突变分别为外显子8 c.2551dupT、外显子8 c.2586delC和外显子9 c.721+1G>A。患儿父母亲均未检测到相应的基因突变。见图4。
五、治疗及转归
11例患儿定期门诊随访,每3~6个月复查。随访时间均超过8个月,最长14年。10例在起病后皮损缓慢增多,起病后8~18个月内皮损稳定,平均生长期为11.3个月。10例生长发育良好,未见其他脏器及肢体发育畸形。1例累及下肢、运动受限的患儿皮损缓慢持续进展,符合进行性骨发育异常。

图3 原发性皮肤骨瘤腹部皮损组织病理

图4 3例原发性皮肤骨瘤患儿及其父母GNAS基因测序图
讨 论
原发性皮肤骨瘤好发于婴幼儿,继发性皮肤骨瘤以青少年及成人多见。原发性皮肤骨瘤临床表现为质硬的皮肤斑块或结节,组织病理显示新生板层骨形成。目前认为原发性皮肤骨瘤的形成与GNAS基因突变有关,该基因编码的调节腺苷酸环化酶活性的G蛋白α亚单位表达上调,导致骨化通路持续活化[6-7]。我们收集了北京儿童医院皮肤科近8年的原发性皮肤骨瘤11例,平均每年新发病例不足2例,提示原发性皮肤骨瘤少见。11例患儿绝大多数出生后6个月内起病,发病前无前驱病变,出生时未见畸形,皮损特点为大小不等的肤色或淡红色质硬的皮肤结节和斑块,伴表皮轻度萎缩。病变多数位于真皮层,部分伴有皮下脂肪层受累,可见膜内成骨,无炎症反应;所有病例均排除了钙、磷代谢异常及自身免疫性疾病引起的异位性皮肤骨化,符合原发性皮肤骨瘤的诊断。本组11例患儿的家系调查均未见相似病例。3例患儿检测外周血GNAS基因,均发现突变,但父母均未见相关突变,另8例因病情稳定不同意GNAS基因筛查。
原发性皮肤骨瘤包括进行性骨化性纤维发育不良(FOP)、进行性骨发育异常(POH)、板层状皮肤骨瘤(PLOC)及Albright遗传性骨营养障碍症(AHO),因各型原发性皮肤骨瘤预后不同,尚需进一步分类。根据患者的发病部位及皮损特点,皮肤骨瘤又可分为孤立性皮肤骨瘤、播散性皮肤骨瘤、PLOC及面部多发性粟粒样皮肤骨瘤[2]。本组病例可分为孤立性皮肤骨瘤3例和播散性皮肤骨瘤8例,或PLOC 10例和POH 1例。
PLOC又称斑块样皮肤骨瘤,患儿临床呈良性表现,无进行性发展,不伴有钙、磷代谢障碍及器官畸形,组织病理为真皮内骨组织形成,可伴有皮下脂肪组织受累。Worret与Burgdorf[8]提出PLOC的诊断要点包括,出生时即发病或1岁以内起病;无骨相关代谢异常;无其他相关骨瘤;发病前无外伤、感染或其他前驱病变。本组10例PLOC患儿生长发育良好,无骨代谢相关激素异常,大多数在1岁后皮损稳定,未见深部进展及影响肢体运动。这些特点均符合PLOC的诊断标准,虽然1例患儿于22个月龄时发病,但皮损特点、组织病理结果及随访转归均符合PLOC,故纳入本研究。亦有文献报道PLOC可在儿童期或成人期发病[9-10]。POH的发病年龄、临床表现及组织病理特点与PLOC有很多重叠,但最重要的区别为POH在儿童期常出现进展,本组1例患儿病情呈进行性发展,累及深部组织,出现运动障碍,符合POH。也有学者认为PLOC为POH的良性型,张筱雁等[11]报道3例POH,其临床表现符合PLOC,但作者将病例归入POH。本组11例原发性皮肤骨瘤仅1例患儿为POH,因此我们认为应保留PLOC名称。由于POH于1994年由Kaplan等[12]首次报道,此后才逐渐被认识,因此对于婴幼儿皮肤骨瘤病例,仍需要长时间定期随访,警惕POH的可能。
不同类型的原发性皮肤骨瘤(如FOP、POH及AHO等)及继发性皮肤骨瘤(钙化性皮肌炎、硬皮病等)需进一步区分。FOP又称骨化性肌炎,可出生后出现皮肤骨瘤,往往进展至深部组织出现运动功能障碍,病理上为软骨内成骨,其发病与ACVR1基因突变有关。大多数FOP患儿常伴有特征性双足大拇趾短小畸形,结合皮损X线平片骨瘤位于深部组织可以确诊[13]。AHO为先天性遗传性疾病,以骨纤维性发育异常、局限性色素沉着斑及性早熟为特征。AHO的皮肤骨瘤表现可以与PLOC类似,也可以像POH一样进展;本文患者均无家族史,中位随访时间42个月,未观察到FOP及AHO临床特征。另外,本病还需与假性甲状旁腺功能减退症鉴别,后者有低血钙、高血磷及甲状旁腺激素水平升高,可兹鉴别。儿童皮肌炎可伴广泛的皮肤钙化及骨化,需要与本病鉴别,鉴别点在于皮肌炎组织病理可见界面改变及炎症细胞浸润,而原发性皮肤骨瘤无此表现,本组资料显示部分病例碱性磷酸酶、乳酸脱氢酶、肌酸激酶及天冬氨酸转氨酶升高,提示骨化活动,而非肌炎表现[2]。
大多数儿童原发性皮肤骨瘤皮损稳定,预后良好,一般不需要积极干预,但有些患儿为POH,需要密切随访、监测功能障碍。目前无有效方法阻止骨化进展。如出现功能障碍,需要联合外科去除损害,改善功能[14]。
利益冲突所有作者均声明不存在利益冲突
