内嵌镧原子的磁性硅纳米线的第一性原理研究
2019-09-17陈朝华翟晓霞王广亮
陈朝华, 翟晓霞, 王广亮, 谢 尊
(1. 石家庄理工职业学院通识教育学院,石家庄 050228;2. 河北师范大学物理科学与信息工程学院新型薄膜材料重点实验室,石家庄 050024)
1 引 言
硅作为半导体工业的主要元素,已经广泛地应用在电脑芯片、微电子器件和超导材料等.同时,电子元器件小型化、微型化的发展趋势也极大地激发人们去探索新型的硅基纳米材料[1].Si类富勒烯笼状团簇的奇异结构和新颖的电磁特性为研发新型的硅基纳米材料开辟了新的途径,因此,Si的笼状结构研究引起人们的广泛关注[2-4].但是由于Si原子sp2杂化的缺失,Si的类富勒烯笼状结构并不能稳定存在[5].以前的很多理论[6-10]和实验[11-14]研究表明,在Si笼中嵌入宿主原子可以明显地提高Si笼的稳定性.过渡金属(TM)掺杂Si笼团簇已经引起研究人员的兴趣,主要由于掺杂TM原子不仅可以稳定Si笼[9,15],而且这种内嵌TM的Si富勒烯表现出新奇的物理化学特性,例如,较大的能隙[16]、较高的磁矩[17]和可调制的极化率[18].为了寻找合适的内嵌原子来稳定Si笼团簇,包括非金属元素[19-23]、TM元素[24-28]、碱金属元素[29-31]和稀土金属(REM)元素[32-34]在内的多种不同元素掺杂的Si笼团簇已经成为近几年的研究热点.Hiura等[13]报道了一个高稳定性的W@Si12团簇,它具有内包W原子的笼状结构且满足18电子规则.Wang等[35]系统研究了3d TM内嵌的Si笼团簇TM@Sin(n= 15, 16),结果显示Ti@Sin(n= 15, 16)具有高度稳定性.Dognon等[36]提议了一系列满足32电子规则的内嵌金属富勒烯笼团簇(An@Si20)n-(An = U, Np, Pu, Am, Cm),结果证实这些结构也是高度稳定的.应用VASP软件包,Guo等[34]预言了一系列稳定的镧系元素(Ln)内嵌Si16富勒烯笼团簇Ln@Si16(Ln = La, Ce, Pr, Nd, Sm, Eu, Gd, Tm, Yb, Lu),计算结果显示不同Ln的自旋磁矩具有可调制性.基于CALYPSO和DFT,张等[37]研究了PdSin(n= 1-15)的几何结构,并预言了笼状Pd@Si12是非常稳定的幻数团簇.此外,在实验方面,Ohara等[38]基于光电子能谱、化学探针和质谱分析法,成功制备了大量的笼状团簇TM@Sin-(TM = Ti, Hf, Mo, W, n = 15, 16). Koyasu等[14]运用阴离子光电子能谱和质谱分析法,证实了高度稳定笼状团簇 M@Si16(M = Sc, Ti, V)已经被成功合成.
内嵌金属原子的Si富勒烯笼状结构可以作为构建新型纳米材料的基本单元,例如,准一维的纳米线(NW)和纳米管(NT).Wang等[39]预言了一类稳定的珍珠项链状的准一维纳米线,它的结构是由一系列Eu@Si20作为基本单元通过彼此连接组装而成.Xu等[40]在实验上成功合成了内嵌V2的十二面体Si富勒烯V2@Si20,并从理论上预言了由V2@Si20组装而成的准一维纳米线(V2@Si20)n.Guo等[41]研究了一系列内嵌TM的氢化硅纳米管TM@H-SiNT (TM = Fe, Co, Cr, Mn),计算结果显示这些SiNT具有半导体特征,TM原子保持较大磁矩.目前关于La@Si16的研究很多,但是关于多个La@Si16组装而成的二聚体La2@Si32以及纳米线La@SiNW的理论研究还未见报道. 在本文中,系统研究了由两个稳定的笼状La@Si16[34,42]组合而成的类管状二聚体La2@Si32团簇以及由La2@Si32作为单体进一步组装而成的一类内嵌La原子的准一维硅纳米线,详细计算并分析了其成键结构和电磁特性,期望本文的研究结果能为新型硅基纳米材料的研发提供一定的理论参考.
2 理论方法
基于密度泛函理论(DFT),在几何优化过程中,完全开放对称性,采用自旋非限制的双数值缀加极化函数DNP[43]基组.对于交换关联相互作用,选取广义梯度近似(GGA)下的Perdew-Wang 1991(PW91)[44]交换关联函数.考虑到La原子的相对论效应,同时也为了节省计算时间,采用包含标量相对论效应的半核赝势(DSPP)[45].电荷转移和自旋磁矩的计算在Mülliken布局分析下实现[46].自洽循环计算的收敛标准设置如下:总能收敛标准为1×10-6Hartree,力常数、原子位移和能量的截断值分别为1×10-3Hartree/Å、1×10-3Å和1×10-5Hartree.对于优化后得到的结构,计算了振动频率和分子动力学(MD)模拟来验证它们的动力学稳定性和热力学稳定性.所有计算都是在量子化学软件包Dmol3[43, 47]中完成的.
为了验证上述计算方案的可靠性,首先对笼状La@Si16进行结构优化,并与以前的研究结果进行对比.计算得到的La@Si16平均结合能(Eb)以及La原子的磁矩分别为3.757 eV和0.268 μB,这与以前的理论结果(3.766 eV和0.281 μB)[42]是一致的,这说明以上计算方案是可靠的.
3 结果与讨论
为了得到二聚体La2@Si32的基态结构,通过不同连接方式对两个La@Si16团簇进行组装,从而构建了一系列可能的几何结构位型,如图1所示.例如,沿着La@Si16的中轴线方向,不旋转(旋转)45度角,连接两个La@Si16的底边四边形,形成4(8)个Si-Si键,得到La2@Si32-a(La2@Si32-c).此外,也考虑了用两个La原子取代Si34内部的两个Si原子,得到La2@Si32-b等6个异构体.通过对比这些异构体的Eb,从而初步确定La2@Si32的最稳定结构是La2@Si32-a.
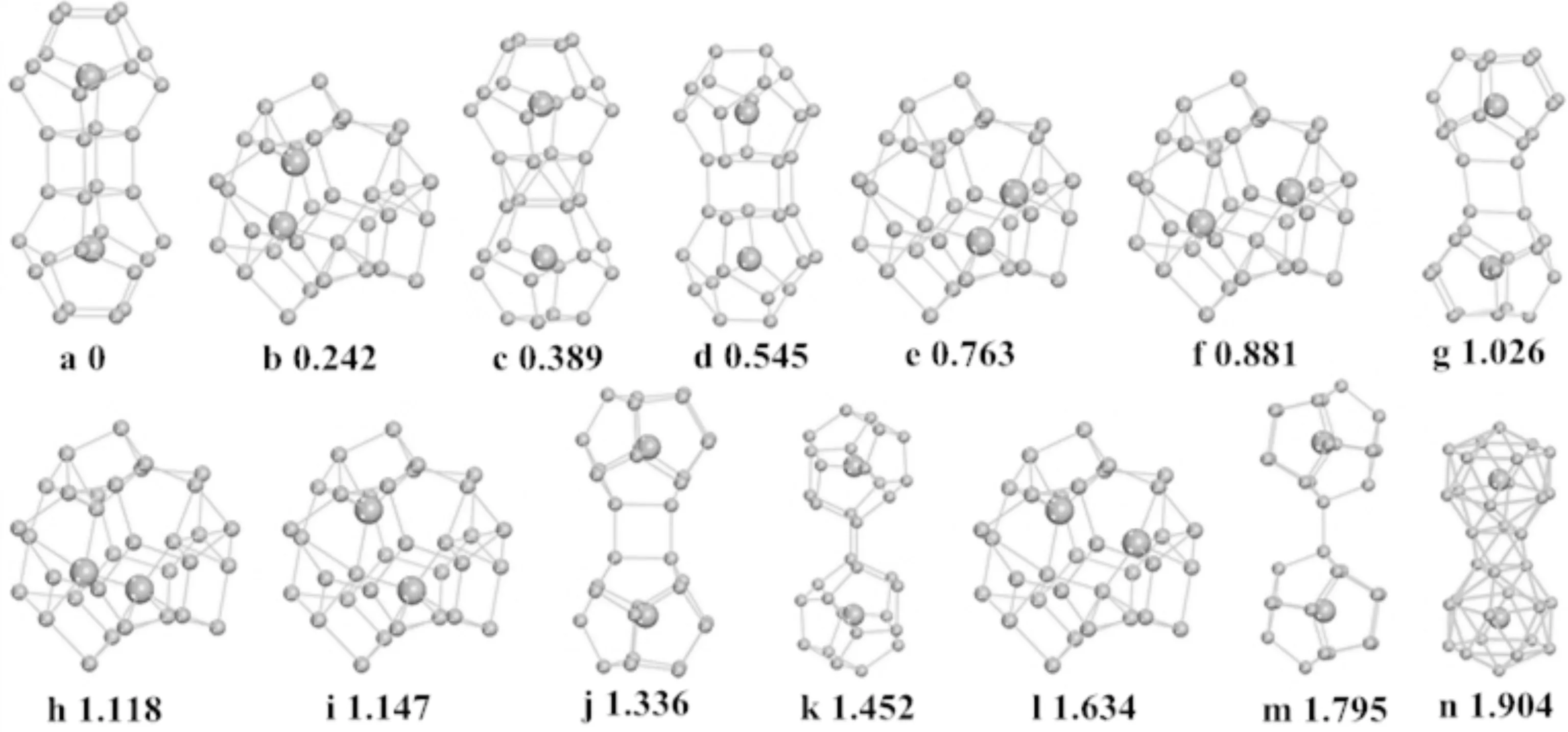
图1 优化后的La2@Si32结构,图片下端为相对于最低能量结构的结合能的差值(eV),大球:La原子;小球:Si原子Fig. 1 Optimized structures of La2@Si32. The binding energy values (eV) beneath each isomer are the relative energies with respect to the lowest lying isomer (La2@Si32-a). Note: large ball, La atom; small ball, Si atom
为了检验La2@Si32-a的稳定性,首先计算了其振动频率.频率分析显示,所有频率中均无虚频出现.其中,最高振动频率和最低振动频率分别为491.2 cm-1和37.7 cm-1,这表明La2@Si32-a具有较高的动力学稳定性.此外,通过开展MD模拟来验证其热力学稳定性.选择在NVE系综下进行MD模拟,初始温度设为1200 K,时间步长和模拟时间分别设为1 fs和1 ps,模拟结果显示,在对应的有效温度约585 K时,La2@Si32-a能够保持原有的拓扑结构,这证明了La2@Si32-a具有良好的热力学稳定性.
为了了解原子之间的成键特性,计算了La2@Si32-a的差分电荷密度(DED),如图2(a)所示.从图中可以看出,成键电子主要聚集在Si-Si之间,显示Si-Si之间的共价键特征.在Si原子周围出现了大量的电子损耗区,而在La原子周围则出现了大量的电子聚集区,表明电荷转移方向是由Si原子转移到了La原子上. Mülliken布局分析显示,Si原子带正电荷,La原子带负电荷,Si原子作为电子的施主,La原子作为电子的受主,平均每个Si原子向每个La原子转移电子为0.148 e,转移的电子主要来源于Si的3s态,这与以上DED分析一致.此外,也计算了管状的Si32(La2@Si32-a移除内部两个La原子之后的结构)的DED,如图2(b)所示,Si原子表现出了类sp3杂化和类sp2杂化,其中在第二、第四、第五和第七层的Si原子呈现出类sp3杂化,其余的Si原子呈现出类sp2杂化.对于第二层和第七层的每个Si原子,四个波瓣中的三个指向最近邻的三个Si原子形成σ键,第四个波瓣指向Si32的外部;对于第四层和第五层的每个Si原子,四个波瓣指向最近邻的四个Si原子形成σ键.对比管状的Si32的DED,La2@Si32-a的DED发生了明显的变化,主要表现在第二层和第七层每个Si原子的类sp3杂化明显减少,而类sp2杂化明显增加,三个波瓣分别指向最近邻的三个Si原子形成σ键.通过以上分析可以看出,由于La原子的嵌入而诱导的类sp2杂化明显地提高了管状Si32的稳定性.此外,计算得到La2@Si32-a的Eb相比较管状Si32的Eb降低了0.134 eV,这也说明了内嵌的两个La原子增加了Si32的稳定性.
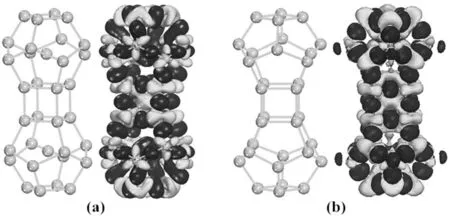
图2 (a)La2@Si32-a和(b)Si32的差分电荷密度Fig. 2 The deformation electron densities of the (a) La2@Si32-a and (b) Si32
为了进一步研究La2@Si32-a的电子结构,计算了前线分子轨道,包括最高占据分子轨道HOMO、HOMO-1、HOMO-2以及最低未占据分子轨道LUMO、LUMO+1、LUMO+2,如图3所示.从图中可以看出,HOMO、HOMO-1和HOMO-2轨道主要定域在Si原子和La原子周围,在La原子上展示出明显的类dxy轨道特征;Si原子之间主要表现出类sp杂化特征,主要形成成键的σ分子轨道;在Si和La之间表现出pd杂化特征,主要形成成键的σ分子轨道.而LUMO、LUMO+1和LUMO+2轨道主要定域在Si原子周围,Si原子之间主要表现出类sp杂化特征,主要形成成键的σ分子轨道.同时,在HOMO和LUMO轨道中,也发现第四和第五层的Si原子之间有少量成键的π分子轨道形成.因此,σ键和π键的形成对于稳定管状结构来说是非常重要的.
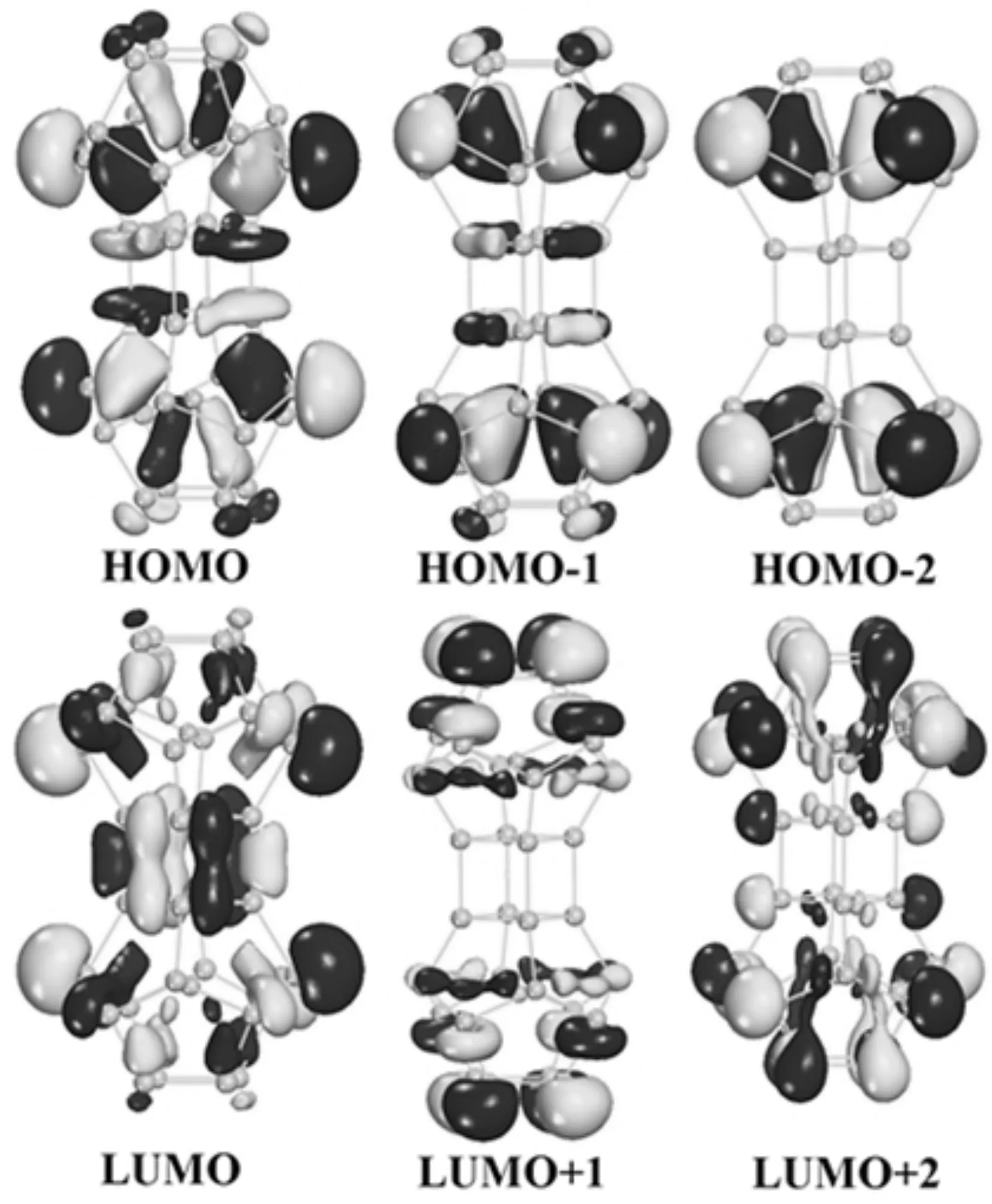
图3 La2@Si32-a的前线分子轨道Fig.3 Selected frontier orbitals for La2@Si32-a
由于团簇的成分、结构和尺寸易于调控,团簇便成为了研究材料磁性的理想媒介.为了研究电荷转移以及磁特性,运用Mülliken布局分析对La2@Si32-a的电荷分布和磁矩做了细节分析.计算结果显示La2@Si32-a的总磁矩为2 μB,每个La原子的局域磁矩是0.172 μB,La原子的5d电子自旋磁矩为0.105 μB,可以看出La原子的局域磁矩主要来源于La原子的5d电子;其余的32个Si原子提供的局域磁矩为1.656 μB,其中第三层和第六层的8个Si原子的总磁矩为1.488 μB,即每个Si原子提供的磁矩为0.186 μB,Si原子的3p电子的自旋磁矩为0.170 μB.由此可见,Si原子的局域磁矩主要是由第三层和第六层的8个Si原子来提供,而每个Si原子的磁矩主要来源于Si原子的3p电子.从分波态密度(DOS)图4(a)上,可以发现DOS主要来自于p态的贡献,而自旋向上的DOS和自旋向下的DOS是不对称的,发生了自旋劈裂现象,因此产生2 μB的磁矩.从自旋密度图4(b)上也可以看出,La2@Si32-a的总磁矩主要来源于第三层和第六层的8个Si原子以及2个La原子的局域磁矩,这8个Si原子以及2个La原子的磁矩平行排列,表现出铁磁性耦合;而第二层和第七层的8个Si原子的磁矩方向与其余Si原子和La原子的磁矩反平行排列,表现出反铁磁性耦合.
在La2@Si32-a中,每个La原子的电子组态是5d3.2766s0.6646p0.227,对比自由La原子的电子组态5d16s2,可以发现6s轨道丢失了一定数量的电子,而5d轨道和6p轨道则得到了一定数量的电子,说明电子在La原子内部发生了转移,La原子内部存在spd杂化.进一步分析发现,6s轨道丢失的电子数(1.336 e)小于5d轨道和6p轨道得到的电子数(2.503 e),说明5d和6p轨道得到的电子除了来源于自身的6s轨道外,还有一部分电子来源于Si原子.通过对Si原子电子组态变化的分析,可知Si原子的3s轨道丢失一部分电子,3p和3d轨道获得一部分电子,在Si原子内部也发生了电荷转移,同样存在着spd杂化.同时,也发现Si原子3s轨道丢失的电子数大于3p和3d轨道获得的电子数,说明Si原子3s轨道除了向自身的3p和3d轨道转移电子外,还有一部分电子转移到了La原子上,这与La原子电子组态的分析是一致的,说明在Si原子和La原子之间也存在着杂化现象.

图4 La2@Si32-a的(a)态密度和(b)自旋密度Fig. 4 The (a) DOS and (b) spin density of the La2@Si32-a
最后,研究了由La2@Si32-a作为基本单元组装而成的周期性的准一维纳米线La@SiNW,如图5(a)所示.优化结果显示,La@SiNW结构中的基本单元La2@Si32-a没有发生明显的扭曲或变形,仍能保持原有的拓扑结构. Mülliken布局分析显示La@SiNW的磁矩是2 μB,每个La原子自旋磁矩为0.007 μB,而所有Si原子的磁矩为1.986 μB,主要来源于第三层和第六层的8个Si原子.La原子磁矩与所有Si原子的磁矩平行排列,表现出铁磁性耦合.为了深入解电子结构,进一步计算了La@SiNW的能带结构以及态密度,如图5(b)和5(c)所示.从态密度图中,发现明显的spd杂化现象,而在费米能级附近有电子态的存在,表明La@SiNW具有金属导电特性.La@SiNW的态密度主要来自于p态的贡献,其次是d态和s态的贡献,而f态的贡献最小.从能带结构图中可以看出,有多条能带穿过费米面,使得费米面具有较大的电子承载能力,可以为电子的可能跃迁提供空位能级.此外,能带在Γ点发生了简并,而且沿着Γ-X轴方向弥散,巡游电子的有效质量较小,因此具有较大的电子迁移率.希望这类磁性二聚体La2@Si32和磁性硅纳米线La@SiNW在自旋电子器件以及高密度磁存储设备方面能够大显身手.

图5 La@SiNW的(a)结构位型,(b)能带结构和(c)态密度Fig. 5 The (a) geometric configuration, (b) band structure and (c) DOS for the La@SiNW
4 结 论
应用第一性原理,系统研究了由两个La@Si16连接而成的管状的二聚体La2@Si32的结构稳定性以及电磁特性.计算结果显示,La2@Si32具有较高的稳定性,主要由于La原子诱导的类sp2杂化提高了团簇的稳定性;Mülliken布局分析表明,La2@Si32的总磁矩为2 μB,主要来源于第三、第六层的Si原子和La原子的局域磁矩;电荷转移方向是由Si原子转向La原子.此外,计算了由一系列二聚体La2@Si32彼此连接组装而成的纳米线La@SiNW的电子结构和磁特性,计算结果显示La@SiNW具有良好的金属导电性质;同时也发现,La@SiNW是具有磁性的,总磁矩为2 μB.期望磁性的二聚体La2@Si32和硅纳米线La@SiNW在未来的高密度磁记录元件和自旋电子器件方面会有广阔的应用前景.
