CH4/H2O/CO2在β-SiO2(100)面吸附的第一性原理研究
2019-09-17赵建飞汪周华
赵建飞, 汪周华, 郭 平, 罗 强
(1. 西南石油大学油气藏地质及开发工程国家重点实验室,成都 610500; 2. 西南石油大学理学院,成都 610500)
1 引 言
作为全球三大非常规天然气之一,页岩气在全世界范围内受到了广泛的重视,美国和加拿大已经实现了页岩气的商业开采[1],并取得较好成果,我国在页岩气的研究现状、成藏机理和有利区评价方面做了大量工作,但近年才开始研究页岩气的吸附现象[2, 3]. 页岩气主要由CH4、C2H6、CO2、N2等气体组成,其中CH4含量可达95%以上[4],其赋存介质为页岩,通常在潮汐沼泽或者深水盆地的水环境下沉积,水充填在有机质孔隙及无机矿物孔隙中[5]. 页岩的矿物组成以石英为主,部分地区石英含量在35.7%~92.6%之间[6]. 根据不同气体的吸附能力不同,许多学者提出页岩气藏注二氧化碳提高采收率[7],因此,研究CH4、CO2和H2O在页岩主要矿物成分SiO2表面的吸附,对了解页岩气的真实赋存状态、定量评估气体的吸附能力、注气开采等方面具有重要意义.
研究者们采用大量的实验和理论研究了页岩气与页岩基质的吸附作用[8-10],研究表明页岩气主要吸附于干酪根和黏土矿物中,很少研究石英表面的吸附. 吉利明等[4]采用等温吸附实验方法,对不同来源和成因的泥页岩中的常见黏土矿物进行了甲烷吸附实验,结果表明不同类型的黏土矿物的气体吸附能力有明显差异,但无直接证据表明气体在石英表面的吸附性,而部分学者研究认为,吸附气量与石英含量呈正相关性[11],故石英表面吸附有待深入研究. 常规油气向页岩的发展其实就是微米空间向纳米空间的发展,宏观上突破越来依赖于微观成果,因而采用新理论方法加强微观研究显得尤为重要[12]. 应用量子力学理论的第一性原理计算是从电子结构出发,只借助于基本常量和合理近似来进行的计算方法,它能够从电子和原子层面认识材料的物理性质,近年来,利用第一性原理在CH4的吸附研究中取得很多有价值的研究成果[13]. 李萍等[14]采用基于密度泛函理论的广义梯度近似方法(GGA/PE91)研究了甲烷在Ni(110)表面不同高对称位的吸附行为,研究结果表明,CH4在Ni(110)表面顶位T4的吸附最稳定,吸附能为4.59 kJ/mol,为物理吸附. Rodríguez-Kessler等[15]用GGA-PBE泛函研究了CH4在最稳定Nin(2-16,21,55)团簇表面的吸附,研究结果表明CH4在Ni4团簇结构的顶位吸附时最稳定,吸附能为-0.47 eV. Zhu等[16]采用DFT-D3的方法研究了甲烷在页岩中干酪根表面的吸附机制,结果表明,甲烷与干酪根之间的相互作用主要是范德瓦尔斯力,其吸附能只有14 kJ/mol左右. 王三跃等[17]采用GGA/PW91泛函研究了甲烷在MOF-5金属有机骨架中不同吸附位吸附的结构模型,并计算了甲烷的吸附能,甲烷的吸附与含氧极性官能团的朝向有关,当碳氢键指向氧位时,甲烷具有最优吸附结构,同时,在骨架中引入电子基团可以增强MOFs对甲烷的吸附作用. Luo等人[18]基于密度泛函理论的第一性原理方法,研究了页岩气在CaCO3(100)面吸附的结构和电子性质,其对致密气在石英砂岩表面吸附性强弱的论证[19]为本研究提供了借鉴. 通过前人的研究发现,在第一性原理计算研究中,从页岩气藏注二氧化碳提高采收率角度以及含水角度出发研究页岩气在其主要无机矿物上的吸附较少,基于此,采用密度泛函理论研究CH4、H2O、CO2在β-SiO2表面不同位置吸附的稳定性,对比分析其吸附状况,进而研究其吸附作用的吸附能、态密度等微观机理.
2 计算方法及过程
2.1 计算方法
使用基于密度泛函理论(Density Function Theory,DFT)[20, 21]的第一性原理计算方法研究CH4、H2O和CO2气体在β-SiO2表面不同位置吸附的几何结构和电子特性,对比分析其在不同高对称位的吸附状况. 计算使用的CASTEP[22, 23]是一种从量子力学角度对固体材料进行理论模拟研究的从头算量子力学程序,由英国剑桥大学凝聚态理论研究组开发. 模拟计算时,采用Perdew等[24]提出的广义梯度近似(Generalized Gradient Approximation,GGA)中的PBE泛函处理交换关联势,同时各收敛精度设为:单原子能量收敛精度2.0×10-5eV/atom,作用于单原子上的最大力收敛精度0.05 eV/Å,最大压力收敛精度0.1 GPa,最大位移收敛精度0.002 Å. 同时,采用Broyden等提出的BFGS优化算法[25-28]进行优化,平面波截止能测试后选取380 eV,SCF自洽精度设为单原子能量收敛至2.0×10-6eV,K点[29]网格尺寸采用1×1×1.
2.2 理论模型
采用Materials Studio 2016数据库提供的SiO2_quartz_beta模型,晶格常数为a=b=5.01 Å,c=5.47 Å,α=β=90°,γ=120°,优化后建立p(2×2)超晶胞作为初始模型并截取(100)面,建立厚度20 Å的真空层,模拟β-SiO2(100)面并对其继续结构优化. 为减少运算量以及弱化基底厚度影响,吸附计算前弛豫表面,即对表面Si原子以外的原子进行固定而表层硅原子与吸附质在结构优化计算中位置可变. 吸附质与表面的距离试算后设为4 Å,在覆盖度θS[30]为0.25ML(monolayer)的条件下对吸附情况进行研究.
β-SiO2(100)面表面层由四个Si原子构成一个矩形,[100]方向边长为5.087 Å,[010]方向边长为5.580 Å. 吸附质分子在β-SiO2(100)面的高对称吸附位有顶位(T位)、长桥位(LB位)、短桥位(SB位)、表面四重洞位(H位),图1为高对称吸附位示意图. 为利用吸附质分子结构的对称性,CH4以碳原子为中心,两个氢原子在下等高,另外两个氢原子在上等高;H2O以氧原子为中心,两个氢原子在下等高;CO2以碳原子为中心三个原子等高且平行于吸附面.
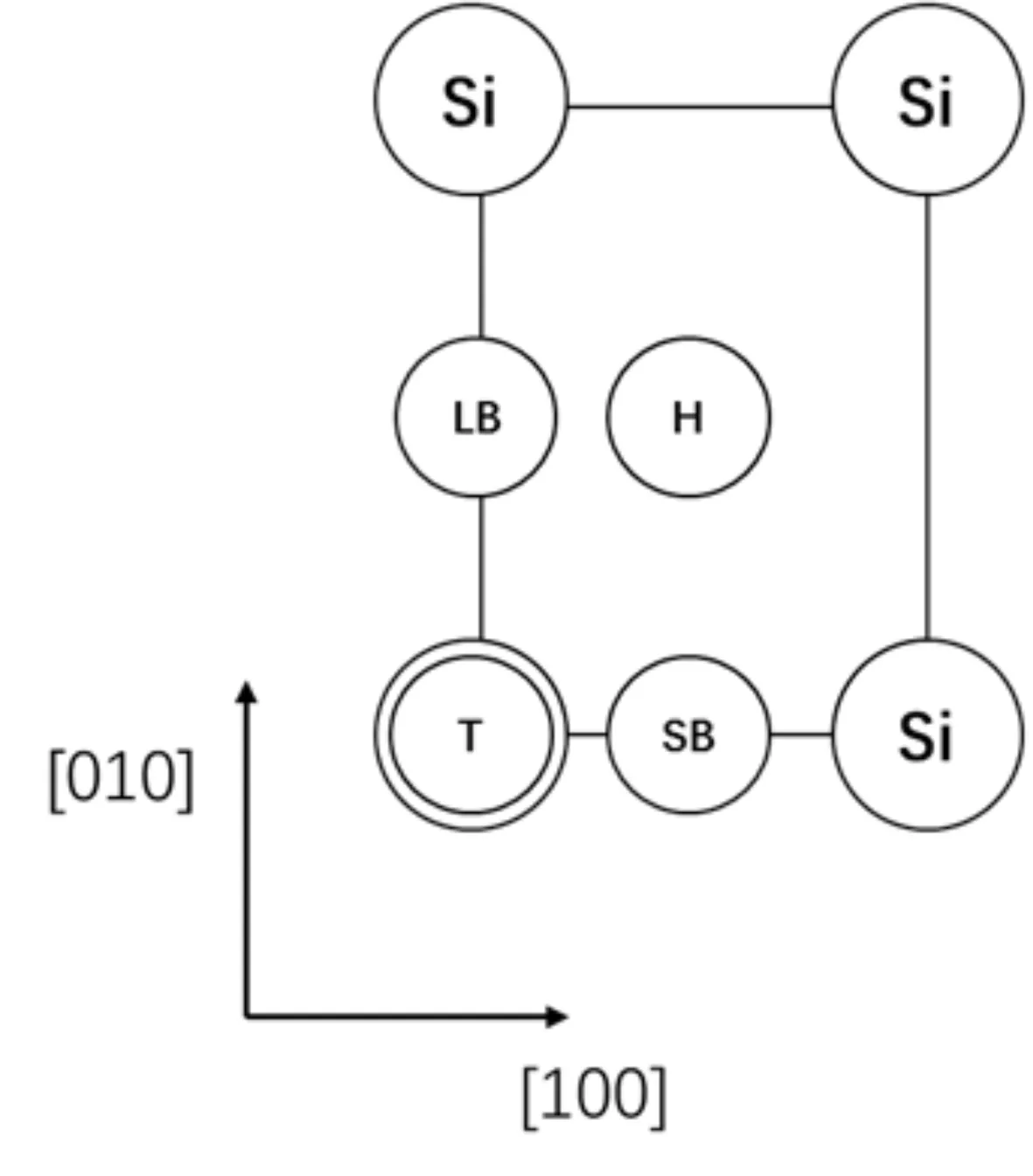
图1 高对称吸附位:T、H、SB与LBFig.1 Adsorption sites: T, H, SB and LB
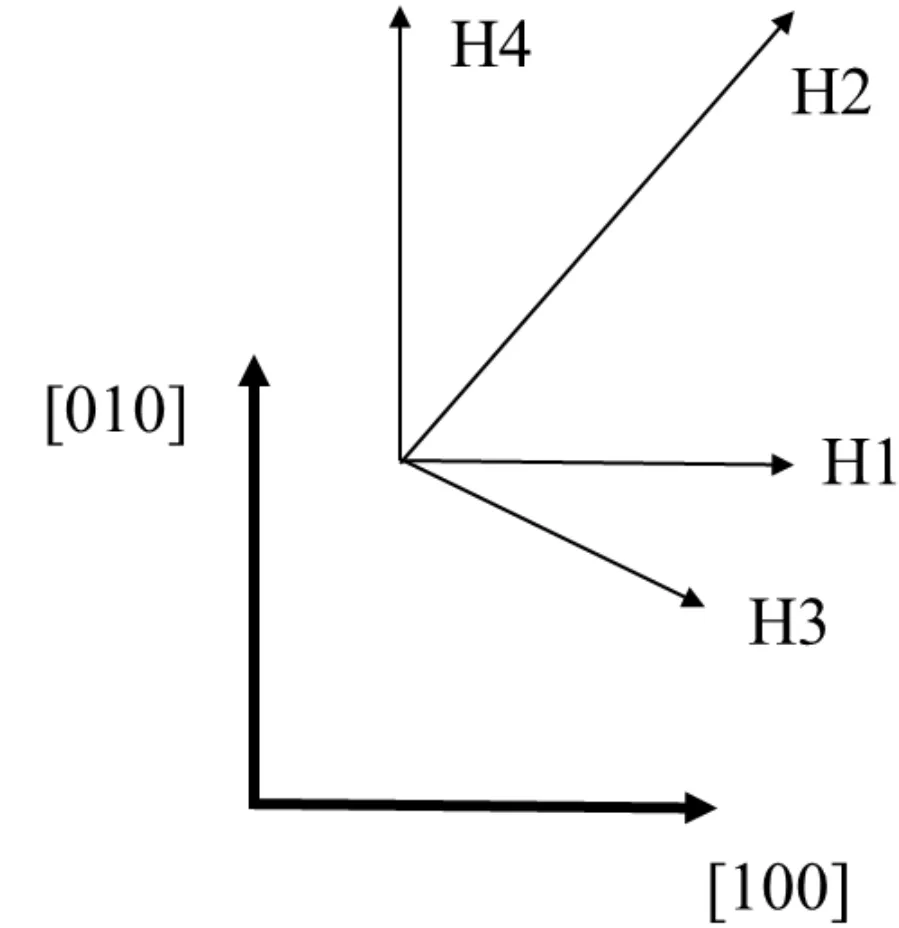
图2 H位四个吸附方向Fig.2 Four adsorption directions of H site
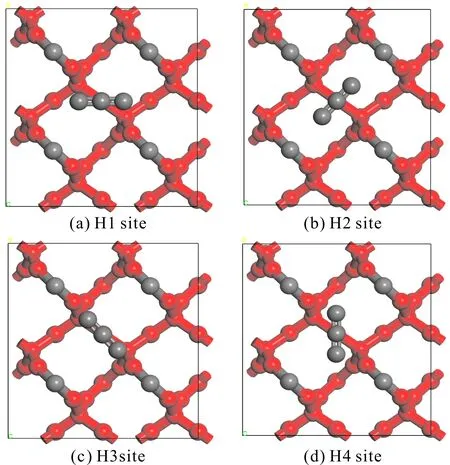
图3 H位吸附方式示意图(红色:被固定的原子,灰色:未被固定的原子)Fig. 3 Adsorption modes of H sites (red: fixed atoms, gray: unfixed atoms)
3 计算结果和分析
3.1 吸附质在β-SiO2(100)面上吸附统计
吸附能表示吸附前后体系总能量的变化,计算公式如下:
Ead=EAdsorbate/SiO2-(EAdsorbate+ESiO2)
(1)
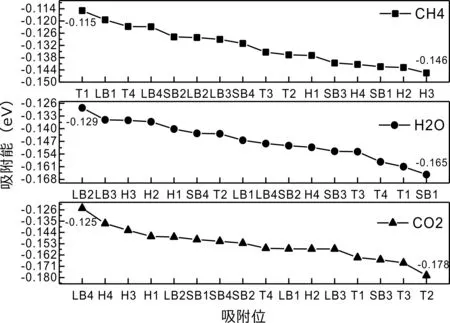
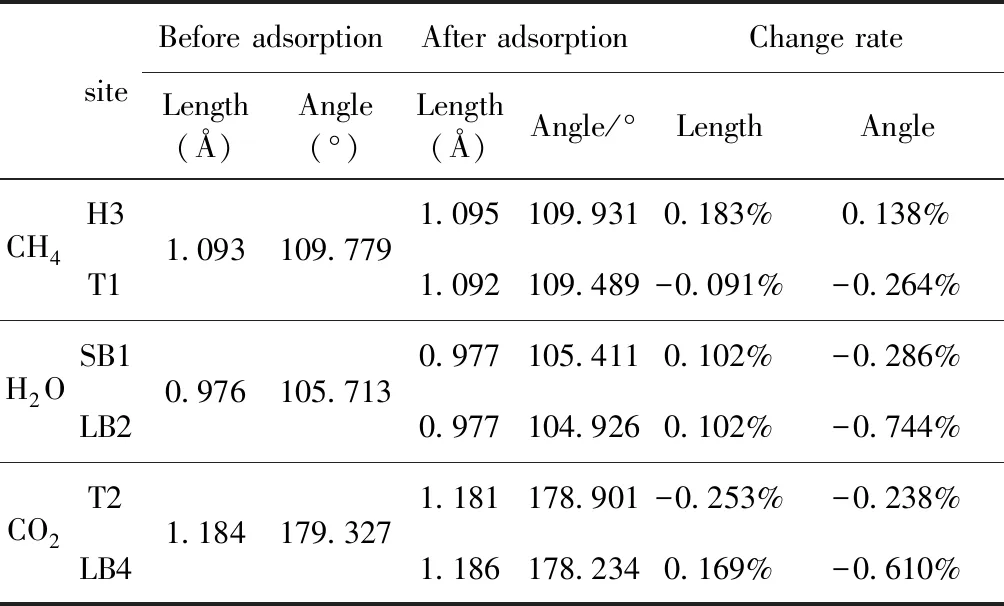
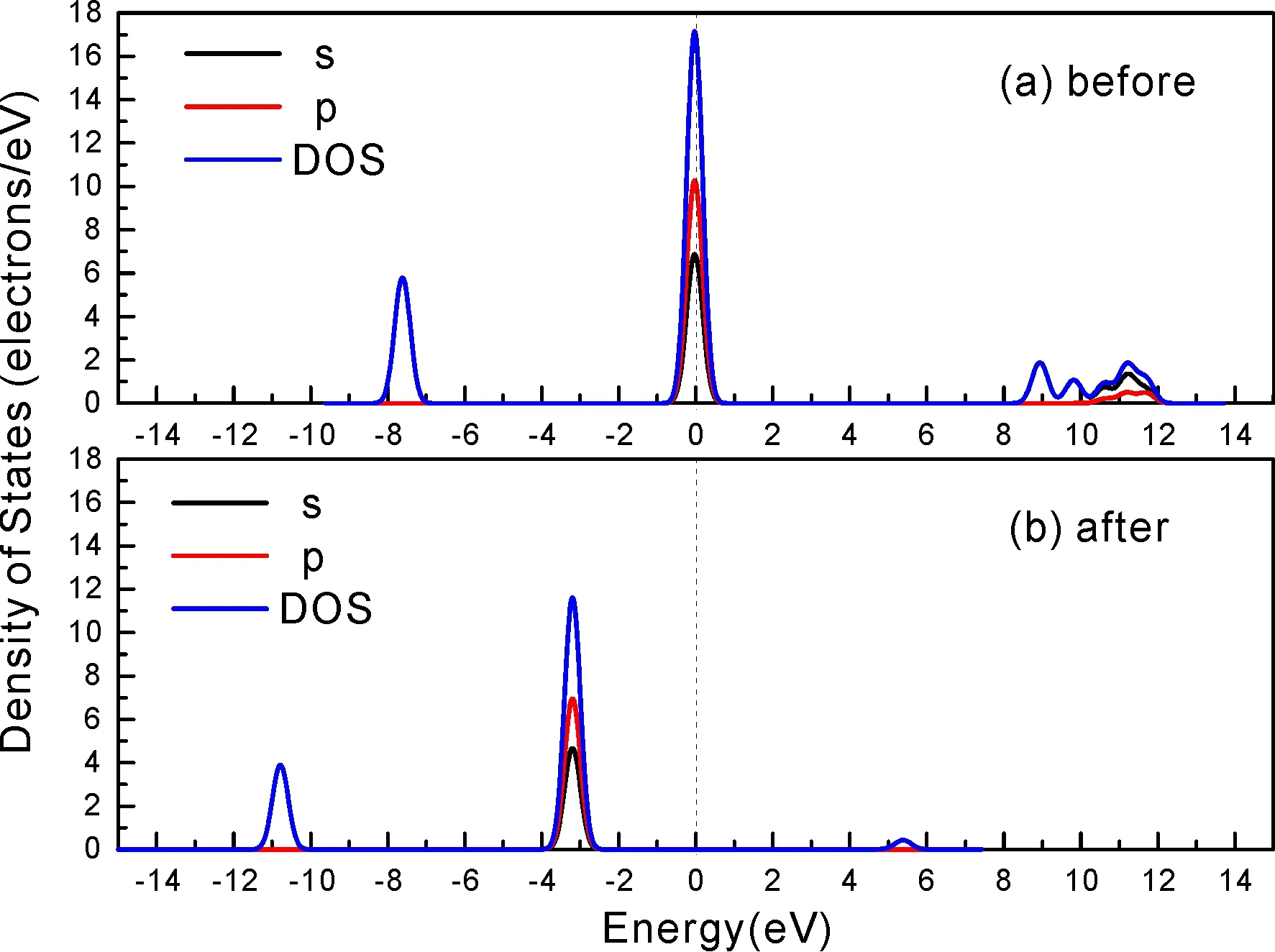
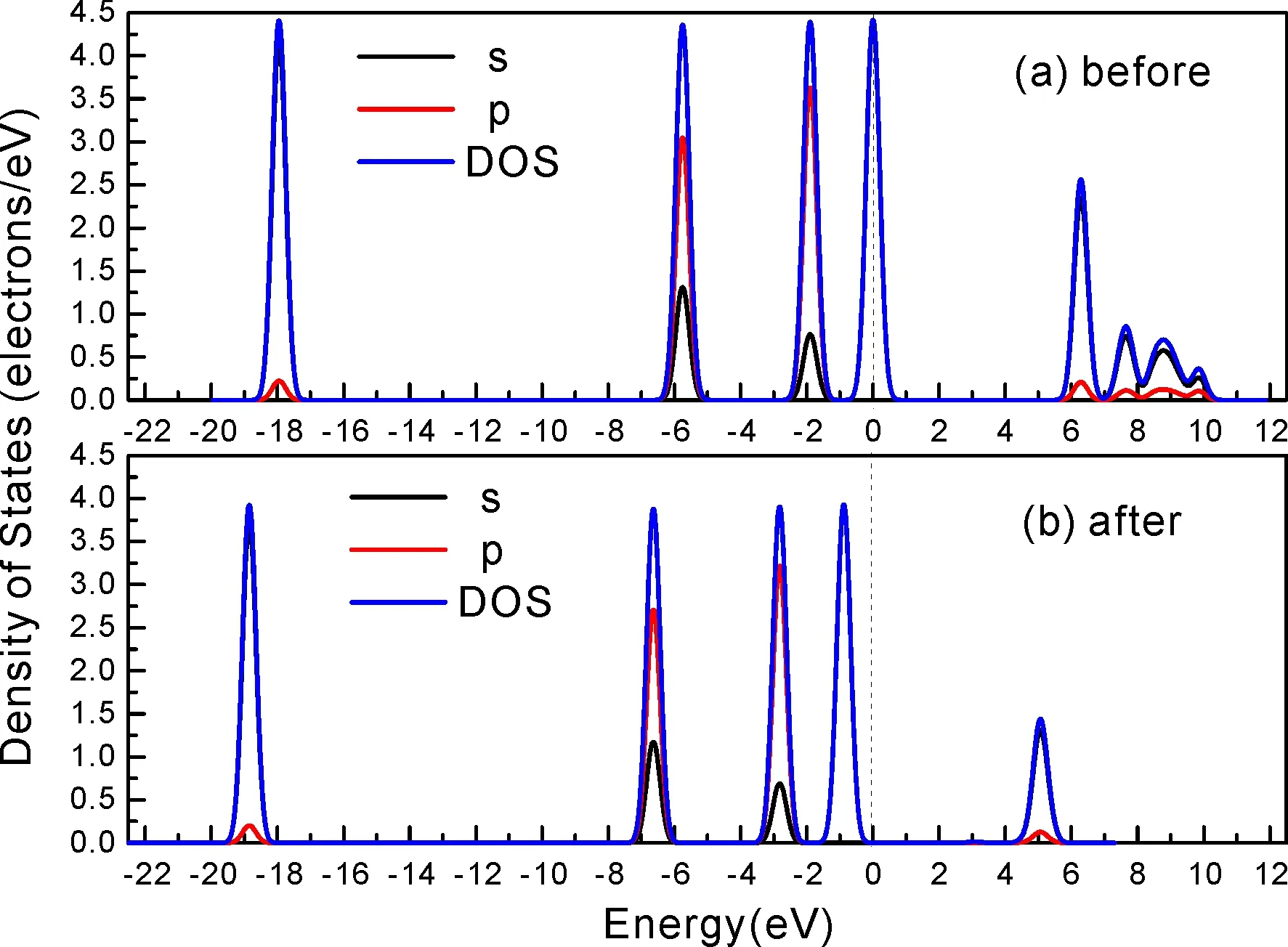
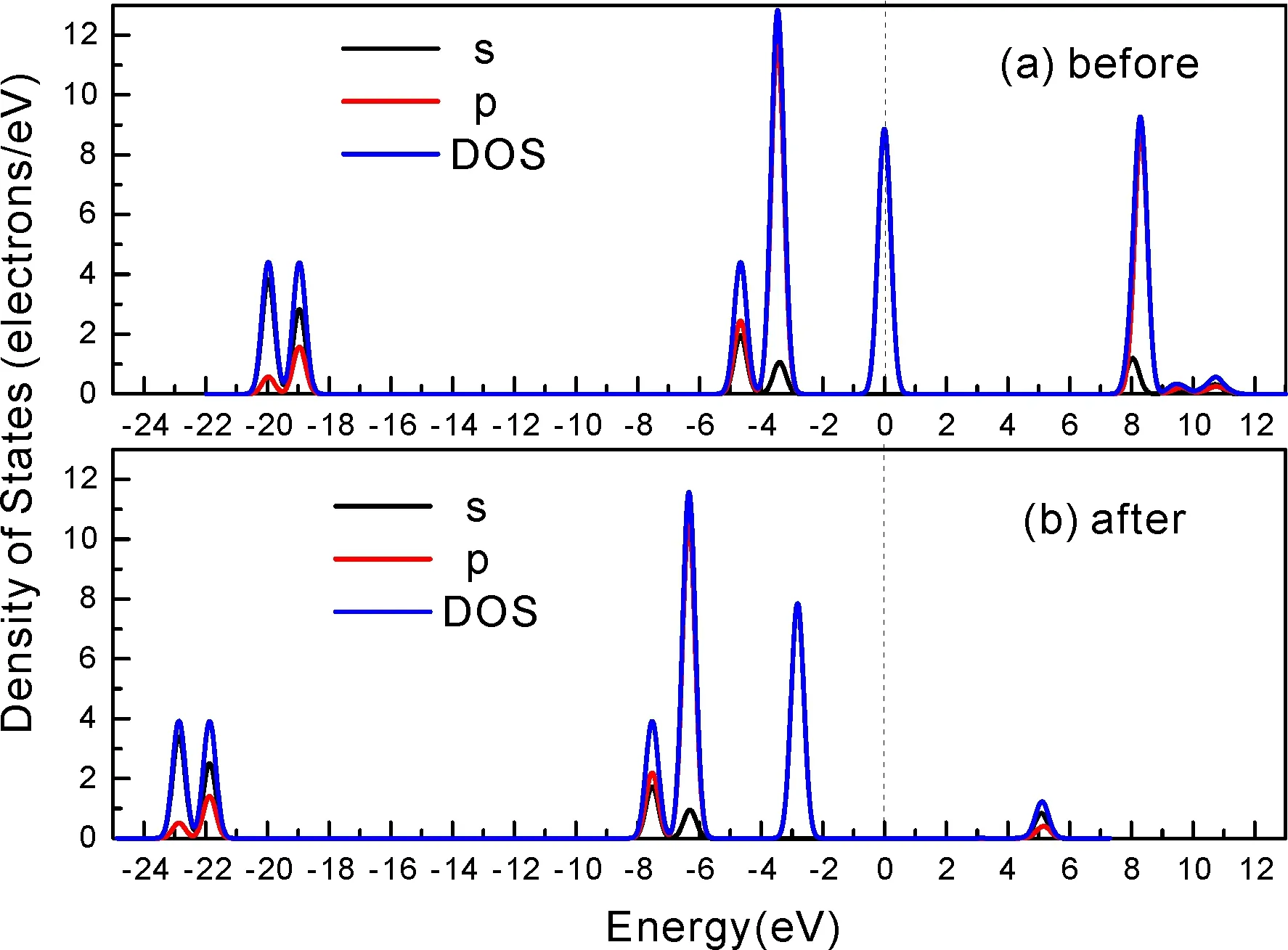
式中,Ead表示各吸附位的吸附能;EAdsorbate/SiO2表示吸附质吸附在β-SiO2(100)面时体系的总能量;EAdsorbate表示各吸附质的能量;ESiO2表示β-SiO2(100)面吸附前的能量. 吸附能计算值为负时表明发生放热反应且吸附后体系更加稳定,Ead越小表示体系的稳定性越强;反之,Ead越大表示体系的稳定性越差. 吸附能满足-0.62 eV 表1 CH4、H2O和CO2在β-SiO2表面的吸附能(eV) Table 1 Adsorption energies (eV) of adsorbate onβ-SiO2(100) surface TEad,HEad,LBEad,SBEad,ⅠT1-0.115 H1-0.137 LB1-0.119 SB1-0.142 T2-0.137 H2-0.143 LB2-0.128 SB2-0.128 T3-0.135 H3-0.146 LB3-0.129 SB3-0.141 T4-0.123 H4-0.141 LB4-0.123 SB4-0.131 ⅡT1-0.161 H1-0.140 LB1-0.146 SB1-0.165 T2-0.143 H2-0.136 LB2-0.129 SB2-0.149 T3-0.153 H3-0.135 LB3-0.135 SB3-0.153 T4-0.158 H4-0.150 LB4-0.148 SB4-0.143 ⅢT1-0.164 H1-0.147 LB1-0.157 SB1-0.150 T2-0.178 H2-0.157 LB2-0.148 SB2-0.153 T3-0.168 H3-0.142 LB3-0.157 SB3-0.166 T4-0.157 H4-0.137 LB4-0.125 SB4-0.151 由表1可知,CH4、H2O和CO2在β-SiO2(100)面不同吸附位的吸附能分布在-0.2 eV ~ -0.1 eV区间内(1 eV=96.4853 kJ/mole[32]),均大于-0.62 eV且小于0 eV,为物理吸附. 根据上表数据做出吸附能随吸附位由大到小排列的关系曲线图,如图4所示. 图4 吸附质在β-SiO2(100)面吸附能随吸附位变化曲线Fig. 4 Adsorption energy curves of adsorbates on the surface of β-SiO2 由图4可直观的看出,CH4在β-SiO2(100)面H3位吸附能最小,为-0.146 eV,表示CH4在该位吸附最稳定,而在T1位吸附能最大,为-0.115 eV,即CH4在T1位吸附最不稳定,吸附能远低于40 kJ/mole,表明CH4在β-SiO2(100)表面的吸附为物理吸附,与文献[33]研究结论一致. H2O在SB1位吸附能最小且为-0.165 eV,而在LB2位吸附能最大且为-0.129 eV. CO2在T2位吸附能最小且为-0.178 eV,而在LB4位吸附能最大,为-0.125 eV. 不同吸附质最小吸附能大小依次为: CH4> H2O> CO2,即,CO2的吸附能力最强,H2O次之,CH4最弱. 同时,各吸附质不同吸附位的吸附能变化范围非常狭窄,最大吸附能与最小吸附能分别相差0.031 eV、0.036 eV、0.053 eV,表明不同吸附位对吸附质分子的吸附影响较小,导致其在表面上容易发生稳定流动[19],但相对而言β-SiO2(100)面优先吸附CO2. 对比分析吸附质吸附前后以及吸附能最大与最小时的几何结构变化有利于理解吸附作用强弱,表2列出了吸附质在最稳定吸附位与最不稳定位吸附前后的物理结构,变化率为正表示键长伸长或键角增大,为负表示键长缩短或键角减小. 由表2可知,CH4在H3位吸附后键长变化率为0.183%,键角变化率为0.138%;而CH4在T1位吸附后键长变化率为-0.091%,键角变化率为-0.264%;H2O在SB1位吸附后键长变化率为0.102%,键角变化率为-0.286%;而在LB2位吸附后键长变化率为0.102%,键角变化率为-0.744%;CO2在T2位吸附后键长变化率为-0.253%,键角变化率为-0.238%;而在LB4位吸附后键长变化率为0.169%,键角变化率为-0.610%. 吸附能最大时吸附质的键角绝对变化率均大于吸附能最小时的,吸附质的物理结构变化微弱表明其所受作用力微弱[14],[34]. 表2 吸附质吸附前后的物理结构 Table 2 Physical structures of adsorbate before and after adsorption siteBefore adsorptionAfter adsorptionChange rateLength(Å)Angle(°)Length(Å)Angle/°LengthAngleCH4H3T11.093109.7791.095109.9310.183%0.138%1.092109.489-0.091%-0.264%H2OSB1LB20.976105.7130.977105.4110.102%-0.286%0.977104.9260.102%-0.744%CO2T2LB41.184179.3271.181178.901-0.253%-0.238%1.186178.2340.169%-0.610% 借助态密度(Density of States,DOS)分析可以进一步理解吸附质在β-SiO2(100)面的吸附作用,分态密度(Partial Density of States,PDOS)可以分析体系吸附后原子的轨道对态密度的贡献,s分态密度由体系各原子不同s轨道杂化构成,p分态密度由p轨道杂化构成. 图5所示为各吸附质最稳定吸附位体系的态密度图,由图可知,态密度曲线在-23.2~ -21.2、-11.1~ -10.3、-7.0~ -6.0、-3.6~ -2.7,-1.1 eV~ -0.4、2.7~ 6.0 eV区间内存在差异,其余能量区间态密度重合. 其中,能量在-23.2 eV~ -21.2 eV区间内态密度为T2位独有,由s分态密度贡献;-7.0 eV~ -6.0 eV区间内T2位态密度均大于H3、SB1位,由p分态密度贡献;-3.6 eV~ -2.7 eV区间内H3位态密度左移,由sp分态密度共同作用,T2位态密度大于SB1位态密度则由p分态密度贡献. 各吸附质最稳定吸附位态密度高度重合表明各吸附质与β-SiO2表面相互作用相似且差异较小. 图5 H3、SB1 与T2位态密度Fig. 5 DOSs of H3、SB1 and T2 sites 图6 (100)面吸附前后态密度Fig. 6 DOSs of (100) surface before and after adsorption 图6为吸附质最稳定吸附位吸附前后基底面的态密度,从图中可以看出,吸附前与吸附后表面的态密度曲线基本重合,仅在能量为3.3~ 3.6、5.1~ 5.7 eV区间有较小差异,表明不同吸附质吸附对β-SiO2(100)表面DOS影响很小. 对比s和p分态密度可以看出,表面的DOS由s、p共同贡献,但各自有明显的界限,低能量区(-21.3 eV~ -18.6 eV)时,s分态密度对DOS起主要作用,高能量区则p对DOS起主要作用,吸附前后PDOS分别与吸附前基本重合,进一步说明吸附作用对表面影响小,吸附质在β-SiO2(100)面的吸附非常微弱,为进一步理解吸附作用对吸附质影响,对CH4、H2O和CO2各自进行吸附前后态密度对比分析. 图7 CH4吸附前后态密度Fig. 7 DOSs of CH4 before and after adsorption 图7为H3位吸附前后CH4态密度,由图可知,CH4的态密度由s态和p态电子贡献. 对比吸附前后态密度分布可知,吸附前态密度分布在-8.1~ -7.0、-0.7~ 0.5与8.4~ 12.1 eV区间,吸附后主要分布在-11.2~ -10.3和-3.8~ -2.8 eV,吸附后CH4的态密度整体向更低能量区域偏移大约3.2 eV且态密度峰值降低,吸附后能量降低结构更加稳定,原8.4 eV~12.1 eV处的态密度几乎消失.s分态密度由吸附前0 eV处的6.875 electrons/ eV,-7.6 eV处的5.785 electrons/eV变为吸附后-3.2 eV处的4.662 electrons/eV,-10.8 eV的3.886 electrons/eV,降幅约2.2 electrons/eV,p分态密度由吸附前0 eV的10.269 electrons/eV变为吸附后-3.2 eV的6.943 electrons/eV,降幅约3.3 electrons/eV. 吸附后电子态密度峰值的数量减少,且峰值均有不同程度的下降,说明吸附作用对CH4的电子态密度的分布有着较显著的影响. 图8 H2O吸附前后态密度Fig. 8 DOSs of H2O before and after adsorption SB1位吸附H2O前后的态密度如图8所示,吸附后H2O的态密度整体向低能量区域偏移约0.8 eV,且态密度峰值由吸附前多个主要峰值降为5个主要峰值,除高能量区变化较为明显外,低能区峰值变化较小,说明吸附后电子更多的占据低能量的能态,吸附后较吸附前的结构更稳定. 图9 CO2吸附前后态密度Fig. 9 DOSs of CO2 before and after adsorption 图9所示为T2位吸附CO2前后的态密度,吸附前态密度分布在-20.6~ -18.5、-5.3~ -3.1、-0.6~ 0.4与7.6~10.8 eV区间内,吸附后主要分布在-23.5~ -21.4、-8.0~ -5.9和-3.7~ -2.1 eV,吸附后态密度曲线整体向低能区移动3.0 eV,峰值具有不同幅度降低,从低能量区到高能量区各峰值分别由4.392、4.402、4.401、12.839、8.856、9.266 electrons/eV变为3.916、3.904、3.913、11.579、7.850、1.224 electrons/eV,其余峰值s与p分态密度近似等比降低. 态密度曲线向低能区移动与态密度峰值降低说明吸附后CO2的电子更多的占据低能量的能态. 此外,对比不同吸附质的态密度图可知,吸附质从CH4、H2O到CO2,态密度分布区间逐渐增大,CO2在能量更低的区域具有态密度分布,与吸附能大小关系相对应,虽然三种吸附质在β-SiO2的吸附为物理吸附,但CO2吸附能更小,更易优先吸附. 基于密度泛函理论第一性原理计算方法研究了CH4、H2O、CO2、β-SiO2(100)面以及吸附质在β-SiO2(100)面吸附的性质,对比吸附质在β-SiO2(100)面上高对称位的吸附能和态密度等特性,得到以下主要结论及认识: (1)CH4、H2O和CO2在β-SiO2(100)面的吸附能分布在-0.2 eV~ -0.1eV区间内,均大于-0.62 eV且小于0 eV,为物理吸附;最小吸附能大小依次为:CH4> H2O> CO2,即,CO2的吸附能力最强,H2O次之,CH4最弱;各吸附质的吸附能变化范围均非常狭窄,表明不同吸附位的吸附影响较小. (2)各吸附质在最稳定吸附位与最不稳定位的物理结构发生了不同程度的键长键角变化,但其键长键角变化均小于1%,吸附能最大对应的吸附位键角绝对变化率均大于吸附能最小对应的吸附位,吸附质的物理结构变化微弱表明其所受作用力微弱. (3)各吸附质处于最稳定吸附位时基底的态密度基本重合,表明各吸附质与β-SiO2表面相互作用相似且差异较小;CH4、H2O、CO2的态密度曲线均向低能量区偏移且峰值出现不同程度降低,吸附后电子更多地占据低能量的能态,吸附后的结构较吸附前更稳定,且CO2在能量更低的区域具有态密度分布,更易优先吸附. 通过吸附能、物理结构与态密度研究从密度泛函理论的角度说明了CH4、H2O、CO2在β-SiO2表面吸附作用的量子力学机理,对应的宏观表现即页岩储层主要矿物石英中含有一定量的吸附气,且可通过注入CO2开采CH4,以上研究内容对揭示CH4、H2O、CO2在石英含量高的页岩中吸附机理具有重要意义.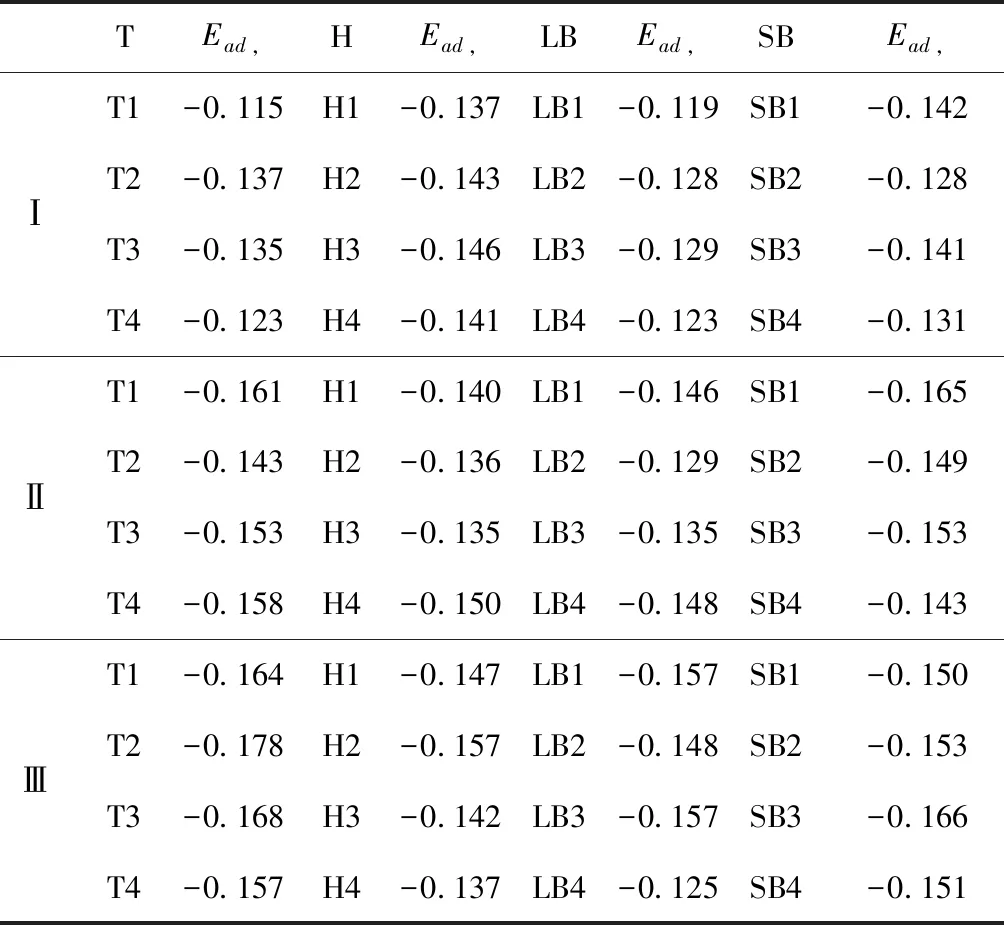
3.2 结构分析
3.3 态密度分析


4 结 论
