硼、氮取代二聚苯中碳原子导致层间相互作用本质变化研究
2019-09-17余兴红郑小玉王一波
余兴红, 陈 颖, 郑小玉, 王一波
(贵州大学化学系 贵州省高性能计算化学重点实验室, 贵阳 550025)
1 引 言
2004年石墨烯(GE)在实验室条件下首次合成出来[1]. 次年,单层六方氮化硼(h-BN)材料也随之被合成出来[2]. GE具有量子磁输运性质、磁性及呈金属特征[3-5],因此GE和h-BN都是具有优异的化学、物理性质的二维(2D)材料,在超级电容器、锂电池以及增加机械阻力等方面的应用较为广泛[6-10]. GE材料经过部分硼、氮取代碳原子形成硼-碳-氮(B-C-N) 2D材料[11,12],而且B-C-N材料是合成多孔GE和h-BN材料的前驱体[13]. 这些2D材料通过van der Waals(vdW)作用形成层间材料[2,14-16],因此也被称之为vdW 2D材料[17]. Li认为h-BN和石墨烯不同的特性与其原子的堆叠型式有关[18],例如,双层GE的Bernal AB堆叠型式改变了电子和光学性能[19-22]. 如图1(Ⅰ, Ⅱ)所示,GE层间以AB型式堆叠[23-25],即碳原子或基本单元苯有平行移位现象. h-BN材料在不考虑硼、氮原子差异的情况下,认为是Bernal AA堆叠型式[25],如图1 (Ⅲ, Ⅳ)所示,既一层h-BN的硼原子对应其上下两层h-BN的氮原子.

图 1 石墨烯和h-BN材料结构图(Å)Fig.1 Graphene and h-BN material structures (Å)
在GE层间材料中其层间相互作用色散作用十分重要[26-30],虽然近年来密度泛函方法发展迅速,但大多数密度泛函方法在不加色散校正的情况下,对色散作用能的预测并不成功,加上经验色散或非局域校正后,计算结果也并不稳定[31,32],CCSD(T)/CBS方法被公认为计算相互作用能最为精确的方法. GE和h-BN层间材料是基本单元六元环无限周期性延展的体系,而B-C-N层间材料存在周期性和非周期性两种,用CCSD(T)/CBS方法直接计算这些材料的层间相互作用,由于计算量太大,目前的算法和计算水平难以实现. 因此选择其基本单元(见图2)的二聚体模型,用CCSD(T)/CBS精确计算其层间相互作用能及其本质是当前间接研究这类二维材料的基础性工作.
前人已用CCSD(T)方法研究过GE基本单元苯的二聚体[33]、h-BN基本单元“无机苯”二聚体[34],其中Viswanathan研究组CCSD(T)/CBS递推采用的是不经BSSE校正的MP2/aug-cc-pVDZ及MP2/aug-cc-pVTZ递推MP2/CBS,CCSD(T)/aug-cc-pVDZ高级项计算也未校正BSSE,显然结果是不可信的[34]. 另外,B-C-N层间材料的基本单元有两种,分别为一对和两对硼、氮取代苯中碳原子[11,12],如图2(Ⅱ, Ⅲ)所示. 一对硼、氮取代苯中碳原子形成的1-氮-2-硼杂苯(BNC4H6)于2009年首次合成出来[35],研究表明BNC4H6具有较大的稳定化能[36]. Korona等人[37]还用CCSD(T)方法研究过BNC4H6与水的作用,但未对BNC4H6二聚体研究. 两对硼、氮取代苯中碳原子有7种同分异构体,Misra在B3LYP/aug-cc-pVDZ水平下计算了稳定化能,结果表明1,3-二氮-2,4-二硼杂苯(B2N2C2H6)的稳定化能最大,甚至比C6H6还大[38]. 为此,本文用CCSD(T)/CBS及SAPT2+方法系统地研究硼、氮取代二聚苯中碳原子的相互作用及其本质,以更好地理解层间材料相互作用本质变化.

图 2 硼、氮原子取代石墨烯基本单元碳原子Fig.2 The boron and nitrogen substituting for carbon in basic unit of graphene
2 计算细节
本文以C6H6,BNC4H6,B2N2C2H6和B3N3H6二聚体为研究对象,从夹心型(S)和平行移位(PD)两种相对位置来建立初始构型. MP2方法配合aug-cc-pVTZ基组对其几何结构优化得到稳定构型. 经过aug-cc-pVTZ和aug-cc-pVQZ两点外推得到MP2/CBS相关能,其中将aug-cc-pVQZ的Hartree-Fock能量看作其极限值. 为了得到更为精确的CCSD(T)/CBS相互作用能,在高级校正项中使用大基组aug-cc-pVTZ. CCSD(T)/CBS相互作用能的计算公式为[39]:
ECCSD(T)/CBS=EMP2/CBS+
(ECCSD(T)/aug-cc-pVTZ-EMP2/aug-cc-pVTZ)
(1)
其中,EMP2/CBS表示在完备基水平下MP2的相互作用能,其表达式为:
(2)
(3)
其中,X,Y分别代表aug-cc-pVTZ和aug-cc-pVQZ,其数值分别为3和4;β=3.05[40].
在SAPT2+/aug-cc-pVDZ水平下对稳定构型的相互作用能进行能量分解. SAPT2+二阶对称匹配微扰理论是在 SAPT2基础上加上对色散能的改进,其静电、诱导、交换项与SAPT2相同,二阶色散校正达到了MP4水平,其表达为[41,42]:
(4)
(5)
(6)
本项研究中的MP2和CCSD(T)方法的工作采用Molpro 2015.1程序[43],SAPT2+对体系能量分解采用PSI4 1.0程序[44],以上全部工作在贵州大学云计算平台上完成的.
3 结果与讨论
为了探索硼、氮取代二聚苯中碳原子所导致层间相互作用的本质变化,主要从S和PD两种相对构型来分析. 在MP2/aug-cc-pVTZ水平下优化了C6H6,BNC4H6,B2N2C2H6和B3N3H6二聚体的几何构型,如图3所示;用两点外推计算了所研究体系的CCSD(T)/CBS相互作用能(见表1). 在SAPT2+/aug-cc-pVDZ水平下分析其相互作用本质,结果列于表2.
3.1 二聚体层间结构分析
如图3所示,C6H6和B3N3H6二聚体存在S和PD构型,BNC4H6和B2N2C2H6二聚体只有PD构型. 根据表1中的ΔECCSD(T)/CBS很容易看出C6H6的PD构型比S构型更稳定;而B3N3H6稳定构型则相反,PD构型不如S构型稳定. 由此可以得出,硼、氮取代二聚苯中碳原子的过程中,其稳定构型从C6H6、BNC4H6和B2N2C2H6二聚体的PD构型向B3N3H6二聚体的S构型转变.
C6H6二聚体的PD构型所对应的堆叠型式为Bernal AB堆叠,在单层GE上放置C6H6、C10H8、C14H10、C14H10、C16H10、C18H12、C20H12、C22H14、C26H16的堆叠型式也为Bernal AB堆叠[27],双层GE亦如此[45]. B3N3H6的S构型所对应的堆叠型式为Bernal AA堆叠,这与h-BN层间Bernal AA堆叠型式一致[23,24,46]. 从表1可以看出,C6H6的PD构型垂直距离D为3.26 Å,这与单层GE和C6H6作用的垂直距离3.3 Å相差较小[26],而且与GE层间距离3.35 Å也仅相差0.09 Å[45]. B3N3H6的S构型D为3.35 Å,这与h-BN材料层间距离3.30-3.33 Å仅相差0.02-0.05 Å[25,47].

图 3 二聚体的S和PD构型Fig.3 The S and PD configurations of the dimers
3.2 二聚体相互作用能
从表1中的CCSD(T)/CBS计算结果可以看到:C6H6的S和PD构型的ΔE分别为-1.36和-2.25 kcal/mol,C6H6的PD构型比S构型稳定;B3N3H6的S和PD构型的ΔE分别为-3.47和-3.11 kcal/mol,B3N3H6的S构型比PD构型稳定;BNC4H6和B2N2C2H6的PD构型的ΔE相近且最大,分别为-4.82和-4.87 kcal/mol,硼、氮取代二聚苯中碳原子后,其ΔE有所增大,尤其是BNC4H6和B2N2C2H6二聚体,同时还可以发现在同一类构型中,相互作用能越大其二聚体两分子间的垂直距离越小.
另外,从表1还可以看到C6H6的S和PD构型的ΔEMP2/CBS分别为-3.45和-5.00 kcal/mol,与ΔECCSD(T)/CBS分别相差2.09和2.75 kcal/mol;B3N3H6的S和PD构型的ΔEMP2/CBS分别为-3.67和-3.26 kcal/mol,与ΔECCSD(T)/CBS分别相差0.20和0.15 kcal/mol;BNC4H6和B2N2C2H6的PD构型的ΔEMP2/CBS分别为-6.60和-5.84 kcal/mol,与ΔECCSD(T)/CBS分别相差1.78和0.97 kcal/mol. 表明MP2/CBS相对于CCSD(T)/CBS相比较而言都过高地估计了相互作用能,而且高估程度不一致. 由此说明本文所研究体系的CCSD(T)/CBS计算,对ΔEMP2/CBS进行高级项校正十分重要.
3.3 二聚体相互作用本质分析
从表2中可以看到,C6H6二聚体的S和PD构型的色散能分别占总吸引能的80%和63%;BNC4H6和B2N2C2H6二聚体PD构型的色散能对总吸引能的贡献相对于C6H6二聚体有所降低,分别为52%和54%;B3N3H6二聚体的S和PD构型的色散能对总吸引能的贡献相对于BNC4H6和B2N2C2H6二聚体的PD构型有所增加,分别为58%和63%. 由此说明在硼、氮取代二聚苯的过程中,其色散能占主要地位,这与萘二聚体[48]、GE与苯[28,29]、苯与萘[49]、双层GE[29]等体系的结果基本一致. 二聚苯以S型堆叠的色散能贡献较PD构型大,而在B3N3H6二聚体中则相反,PD构型的色散能贡献较S构型大.
从表2还可以看出,苯二聚体的S和PD构型的静电能分别占总吸引能的16%和29%,这与体系的堆叠型式密切相关. 分析图4可以得到,在苯二聚体S构型中,正电势、负电势正好相对,静电排斥较大,静电能对总吸引能的贡献较小,仅为16%. 而在苯二聚体PD构型中,两个苯分子间发生了平行移位,正、负电势的区域较好互补,其静电能的贡献增加到了29%. 在BNC4H6和B2N2C2H6二聚体的PD构型中静电势互补程度较高,其静电能贡献分别达到了38%和37%. B3N3H6二聚体S构型的静电能对总吸引能的贡献也为37%,而PD构型则有所下降,为32%,这是由于B3N3H6二聚体以S构型堆叠时,硼、氮原子交叉重叠,能使正、负电势的区域最大程度互补,而PD构型的互补程度不及S构型. 由此可见,静电作用在硼、氮取代二聚苯的体系在一定程度上也起着重要作用;另外,在本文所研究的体系中,诱导作用的贡献相对较小,均在10%以下,诱导作用的差异性与六元环的极化程度相关.

表1 二聚体稳定构型的结构参数和相互作用能
aDandRrepresent the vertical and horizontal distances of the configuration of Fig. 3, respectively.
表2 SAPT2+/aug-cc-pVDZ分解硼、氮原子取代二聚苯中碳原子的相互作用能(kcal/mol)
Table2 Interaction energies (in kcal/mol) at SAPT2+/aug-cc-pVDZ level for the carbon of dimerized benzene replaced by the decomposed boron and nitrogen
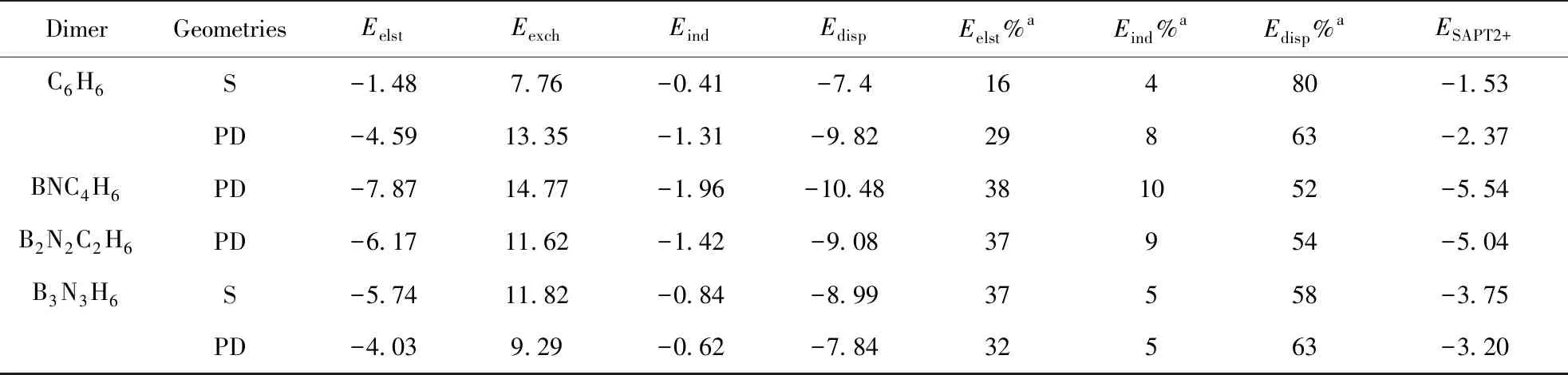
DimerGeometriesEelstEexchEindEdispEelst%aEind% aEdisp% aESAPT2+C6H6S-1.487.76-0.41-7.416480-1.53PD-4.5913.35-1.31-9.8229863-2.37BNC4H6PD-7.8714.77-1.96-10.48381052-5.54B2N2C2H6PD-6.1711.62-1.42-9.0837954-5.04B3N3H6S-5.7411.82-0.84-8.9937558-3.75PD-4.039.29-0.62-7.8432563-3.20
aContribution to the total attractive interactions.

图 4 单分子静电势图Fig.4 The electrostatic potential diagrams of single molecules
4 结 论
本文使用CCSD(T)/CBS计算了硼、氮取代二聚苯中碳原子的相互作用能,在SAPT2+/aug-cc-pVDZ水平下分解了相互作用能,探索了硼、氮取代二聚苯中碳原子所导致层间结构、相互作用能以及层间相互作用本质变化. 得出结论如下:首先、稳定构型从C6H6,BNC4H6和B2N2C2H6二聚体的 PD构型向B3N3H6二聚体的S构型转变,其中,二聚苯的PD构型和B3N3H6二聚体的S构型分别与GE层间材料的Bernal AB堆叠型式和h-BN层间材料的Bernal AA堆叠型式一致;其次、硼、氮取代二聚苯中碳原子后使其层间相互作用能增大,而BNC4H6和B2N2C2H6二聚体较为明显. 通过相互作用能成分的分解分析,发现色散能对总吸引能的作用占主要地位,静电能的贡献次之,诱导能贡献相对较小,硼、氮取代二聚苯中的碳原子后,静电作用的贡献呈现明显增加.
