肉桂酸植物甾醇酯的酶法合成及对DPPH·的清除作用
2019-09-11陈林林荆荣华辛嘉英
陈林林 荆荣华 李 伟 韩 可 辛嘉英,2
(哈尔滨商业大学省高校食品科学与工程重点实验室1,哈尔滨 150076) (中国科学院兰州化学物理研究所羰基合成与选择氧化国家重点实验室2,兰州 730000)
植物甾醇又称植物固醇[1],是化学结构与胆固醇类似的一类天然活性物质,可有效减少人体对低密度脂蛋白胆固醇的吸收、促进胆固醇的降解代谢、抑制胆固醇的合成等[2-5],具有抗癌、抗动脉粥样硬化和抗氧化等功效[6-8]。然而,植物甾醇熔点较高,常温下为结晶形式,在人体中的溶解性和生物可利用性较差;不溶于水,在油脂中的溶解度也很小[9],在食品中的应用受到限制[10]。经研究发现,植物甾醇酯作为植物甾醇的衍生物,具有和植物甾醇相同甚至更优的生理活性功能[11],有效增加其脂溶性和生物利用度[8],逐渐成为研究热点[12]。
植物甾醇酯的制取常采用化学合成法、酶催化合成法及离子液体催化合成法。酶催化合成法同化学合成法相比较具有催化反应条件较温和,副产物较少及易于产物的分离纯化等优点,但需要选择合适的有机溶剂同时提高酶的稳定性,提高生产效率[13]。Nikolaus等[14]发现,和甾醇酯通过脂肪酶催化植物甾醇与脂肪酸酯或三酰甘油酯交换,可有效地制备植物甾醇酯。霍玉洁等[15]设计合成4 种离子液体催化肉桂酸和植物甾醇酯化制备肉桂酸植物甾醇酯,但离子液体的制备工艺较复杂,成本较高,反应体系粘度高、关于其对产物的毒性难以确定,并且关于离子液体催化合成植物甾醇酯的报道较少,研究较分散。基于以上原因,选择安全无毒的生物酶用于合成植物甾醇酯。本研究将优化非水反应体系溶剂,通过脂肪酶催化转酯反应合成肉桂酸植物甾醇酯,构建并优化肉桂酸乙酯与植物甾醇的转酯化反应体系,旨在提高植物甾醇的溶解性、稳定性和抗氧化性,合成具有植物甾醇功效的功能性酯,为甾醇的安全、高效合成提供技术支持。
1 材料与方法
1.1 材料与试剂
固定化南极假丝酵母脂肪酶Novozym435脂肪酶(100 000 U/g)、猪胰脂肪酶PPL(80 000 U/g)、Lipozyme TL IM(100 000 U/g)、Lipozyme RM IM(70 000 U/g)、扩展青霉脂肪酶(70 000 U/g)、脂肪酶LBK4000(70 000 U/g);肉桂酸乙酯、肉桂酸甲酯;植物甾醇(纯度≥90%);4 Å分子筛;甲醇(色谱纯);二氯甲烷、异辛烷、叔戊醇、正己烷、2-甲基四氢呋喃、叔丁醇、四氢呋喃、乙酸乙酯、石油醚、冰醋酸均为分析纯。
1.2 主要仪器
BS210S电子天平;IKA磁力搅拌器;KH-2000型薄层色谱扫描仪;DHG-9203A型电热恒温鼓风干燥箱;R-1001N型旋转蒸发仪;SHB-Ⅳ双A循环水式多用真空泵;U3000高效液相色谱仪。
1.3 实验方法
1.3.1 肉桂酸植物甾醇酯的合成
在15 mL反应瓶中按底物比分别加入0.5 mmol肉桂酸乙酯与植物甾醇、0.5 g分子筛,再加入5.0 mL有机溶剂和一定量的Novozym435脂肪酶,在在一定温度条件下,磁力搅拌器转速300 r/min反应120 h。定时取样分析,计算肉桂酸甾醇酯的产率,反应初速度。分别对酰基供体、脂肪酶种类进行筛选,并考察脂肪酶添加量、温度、底物比等对转酯反应的影响。具体实验条件由反应体系因素的不同有所改变。
1.3.2 产物分离与检测
采用薄层层析(TLC)分析每隔24 h吸取2.5 μL的反应混合物进行反应进程监测。将反应样品和对照样品分别在GF 254硅胶板上点样,展开剂根据筛选的最佳溶剂组分[9]之间的配比为石油醚∶乙酸乙酯(10∶1),展开距离为10 cm,254 nm紫外光下观察,对产物肉桂酸植物甾醇酯、底物肉桂酸乙酯、植物甾醇定性检测。根据薄层层析的展开条件和分离效果,对反应后滤去酶的反应液进行硅胶柱分离纯化,硅胶柱(40 cm×5 cm),按照洗脱剂为石油醚∶乙酸乙酯(15∶1)洗脱,收集各部分洗脱液旋转蒸发后可得到目标产物肉桂酸植物甾醇酯。
.nbhv利用高效液相色谱(HPLC)进行定量分析,色谱柱:XDB-C18(4.6 mm×150 mm, 5 μm);流动相:溶剂A(100% 甲醇)和溶剂B(0.5% 冰乙酸);流速为1 mL/min,保留时间为23 min;样品经甲醇稀释20倍,进样量10 μL;检测波长:278 nm;检测温度:30 ℃。
肉桂酯乙酯产率及各产物产率按各物质峰面积(A)归一化法[16]进行计算:

1.3.3 肉桂酸植物甾醇酯清除DPPH·清除能力测定
吸取浓度为0.025、0.05、0.10、0.15、0.20、0.25 mg/mL的样品乙醇溶液,加入到2 mL浓度为0.16 mmol/L的DPPH乙醇溶液中,充分混合后,在黑暗处室温放置30 min,在517 nm波长下测定吸光度值。空白溶液由1 mL乙醇和2 mL 0.16 mmol/L的DPPH乙醇溶液组成,按公式计算清除率[17],并计算半数清除率浓度IC50值。

式中:A1为样品DPPH混合溶液的吸光度值;A2为样品吸光度值;A空白为空白的吸光度值。
2 结果与分析
2.1 反应产物分析
2.1.1 TLC对反应产物的分析
对肉桂酸乙酯与植物甾醇反应液薄层层析板进行扫描,其薄层扫描结果如图1所示。

注:A—植物甾醇;B—肉桂酸乙酯;C—反应液;D—肉桂酸植物甾醇酯。图1 肉桂酸甾醇酯染色前后TLC示意图
由图1a可以清晰地看出,由Novozym435脂肪酶催化肉桂酸乙酯与植物甾醇反应有4种物质的展开点,通过对照肉桂酸乙酯标的展开点对照可知,Ⅰ点为底物肉桂酸乙酯,比移值为0.82,根据反应机理,理论上产物结构分析Ⅱ处的展开点推测为肉桂酸植物甾醇酯,测定得到比移值为0.31。由图1b可知,A为植物甾醇,因为植物甾醇紫外吸收弱,所以在图1a中并不能显现出来,需要染色后才可以看出。在图1a中可看出,产物肉桂酸植物甾醇酯在紫外显示下明显,经染色后同样可以清晰显示,所以确定图1a中Ⅲ点应为肉桂酸植物甾醇酯。
2.1.2 HPLC对反应产物的分析
使用高效液相色谱仪对底物、反应液与纯化后产物进行测定,得到液相色谱图如图2~图4所示。
在TLC分析结果基础上,进一步通过高效液相色谱定性、定量分析,对应图2中底物肉桂酸与肉桂酸乙酯的保留时间,且由于肉桂酸甾醇酯的极性大于肉桂酸乙酯的极性,故肉桂酸甾醇酯的保留时间小于肉桂酸乙酯的保留时间。因此,图3转酯化反应液中底物肉桂酸乙酯的保留时间为12.050 min,猜测2号峰为产物峰。通过进一步对产物肉桂酸甾醇酯的分离纯化后进行高效液相色谱分析,液相色谱图如图4所示,得到产物的出峰位置,与反应液混点液相色谱图上的吸收峰和保留时间对照,并从从疏水性的大小可判断,图3中保留时间为11.29 min的吸收峰为肉桂酸甾醇酯。

图2 肉桂酸乙酯高效液相色谱图

注:1代表肉桂酸乙酯;2代表肉桂酸甾醇酯;3代表肉桂酸。图3 肉桂酸植物甾醇酯反应液高效液相色谱图
图4 纯化后肉桂酸甾醇酯高效液相色谱图
2.2 非水相体系催化转酯反应条件的优化
2.2.1 酰基供体的筛选
以肉桂酸植物甾醇酯的产率和反应初速度为指标,考察不同底物生成肉桂酸植物甾醇酯的效果,结果如图5所示。
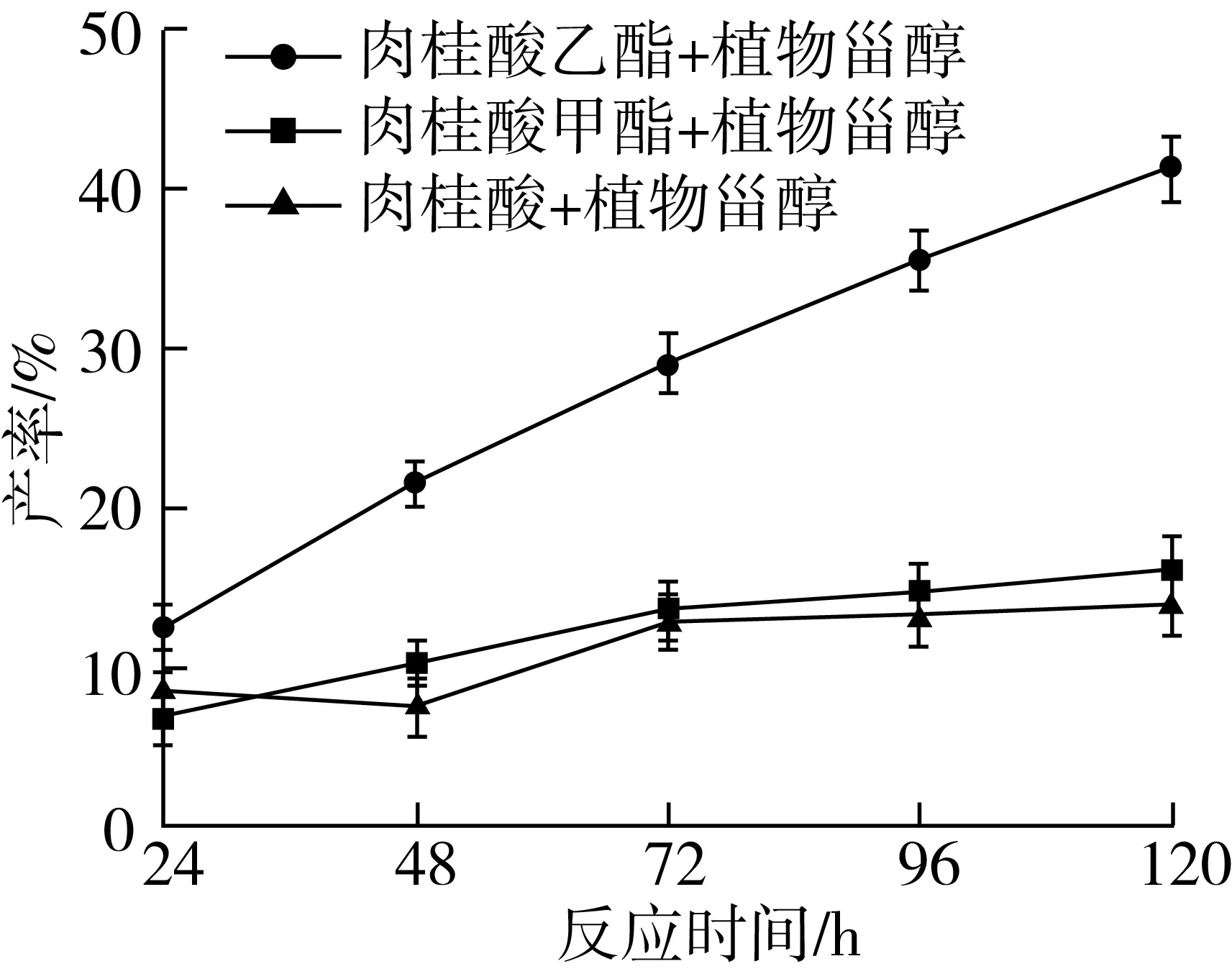
注:反应条件为叔戊醇与2-甲基四氢呋喃混合溶剂(1∶1)5.0 mL,Novozym435脂肪酶80 mg,磁力搅拌器转速300 r/min,反应温度60 ℃,反应时间120 h。图5 不同酰基供体对转酯反应的影响
从图5中可知,随着酰基供体的改变,产率在其他条件相同的情况下有显著变化(P<0.05),肉桂酸乙酯与植物甾醇合成肉桂酸植物甾醇酯的产率明显高于肉桂酸和肉桂酸甲酯作为酰基供体时合成肉桂酸植物甾醇酯的产率,在合成肉桂酸植物甾醇酯的过程中,肉桂酸甲酯水解成肉桂酸的水解率明显高于肉桂酸乙酯。与其他两种酰基供体相比,肉桂酸乙酯在反应前48 h产率增加明显,说明其在反应初速度较高。若用肉桂酸作为酰基供体,虽然可以与植物甾醇直接酯化,而且不存在底物水解的问题,但是产率并不高,反应初速度也很低。因此,选用肉桂酸乙酯作为酰基供体。
2.2.2 脂肪酶种类的筛选
在叔戊醇和2-甲基四氢呋喃混合有机溶剂体系中,选择了六种不同的酶作为催化剂,通过薄层层析未检测到由扩展青霉和LBK4000脂肪酶催化的反应有产物生成,故只需在RM IM、Novozym435、TL IM、PPL这四种脂肪酶中进行筛选,结果如图6、图7所示。
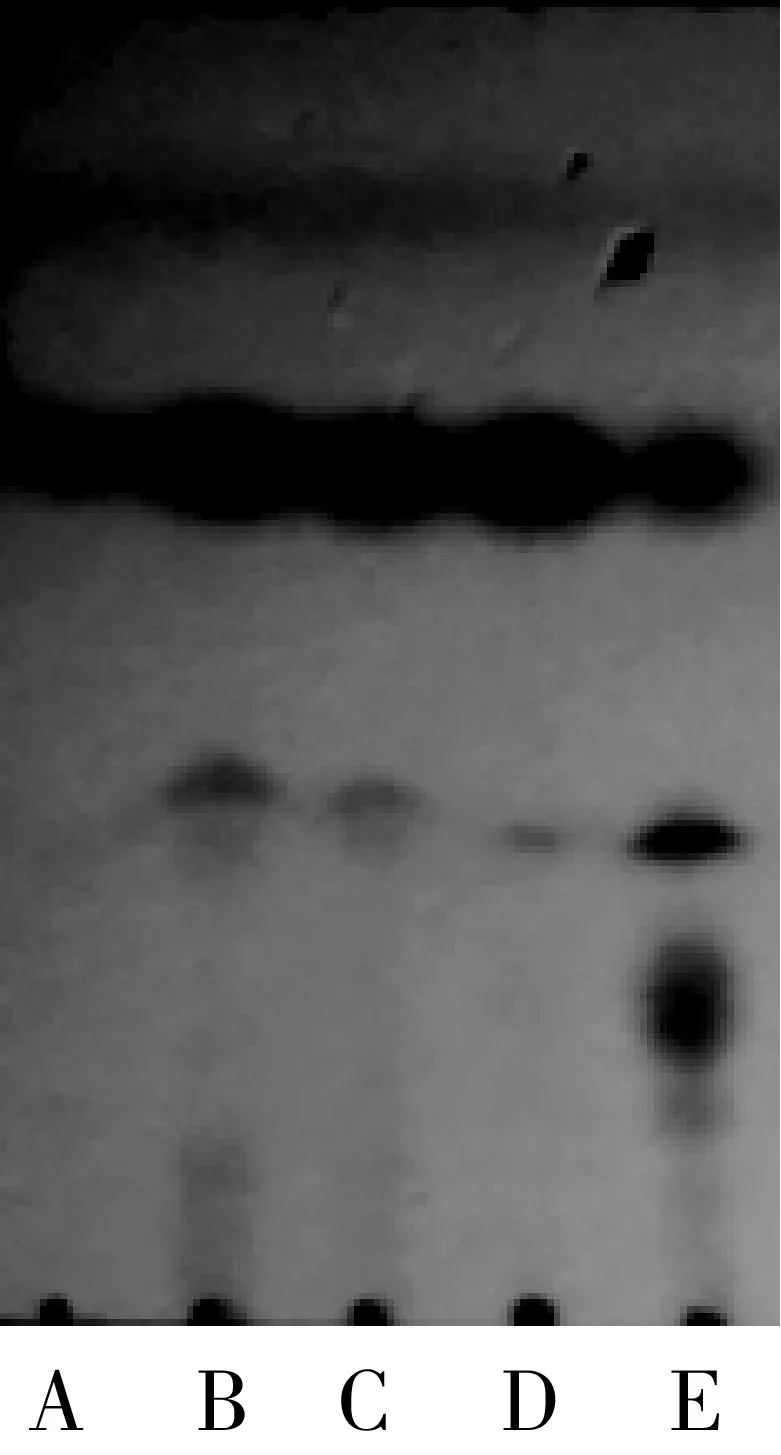
注:A—肉桂酸乙酯;B—PPL酶反应液;C—TL IM酶反应液;D—RM IM酶反应液;E—Novozym435酶反应液。图6 脂肪酶种类筛选反应TCL示意图
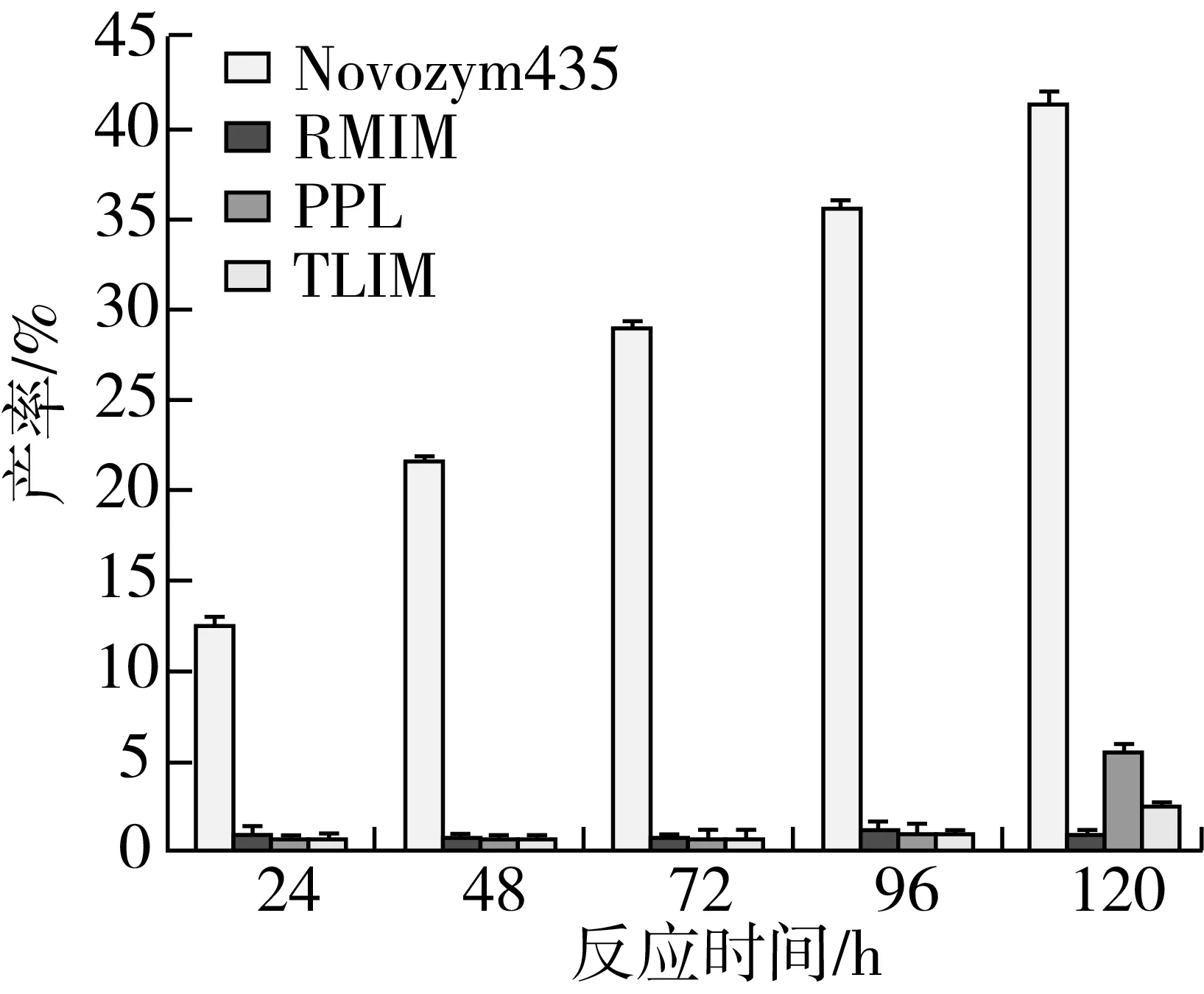
注:反应条件为叔戊醇与2-甲基四氢呋喃混合溶剂(1∶1)5.0 mL,肉桂酸乙酯0.5 mmol及植物甾醇0.5 mmol,磁力搅拌器转速300 r/min,反应温度60 ℃,反应时间120 h。图7 脂肪酶种类对转酯反应的影响
由图7可知,在其他条件相同的情况下,脂肪酶种类的不同,产率差异明显(P<0.05),由Novozym435脂肪酶催化合成肉桂酸植物甾醇酯的产率明显高于其他三种脂肪酶,在120 h时产率达到最大,可达41.29%。与其他种类酶相比,Novozym435脂肪酶在反应初期产率变化明显,说明其催化合成肉桂酸植物甾醇酯的反应初速度较高。并且通过图6可以看出,Novozym435脂肪酶TCL检测获得的产物点最为明显。因此,选用Novozym435脂肪酶进行后续的实验,这个结果与李江涛等[18]研究脂肪酶催化合成α-亚麻酸植物甾醇酯脂肪酶选择的结果一致。
2.2.3 有机溶剂的筛选
分别选用叔戊醇与2-甲基四氢呋喃混合溶剂(1∶1)、异辛烷、2-甲基四氢呋喃、四氢呋喃、叔戊醇、正己烷作溶剂。通过薄层层析检测到在叔戊醇、异辛烷、正己烷作反应溶剂时并没有产物生成,其他溶剂体系结果如图8所示。
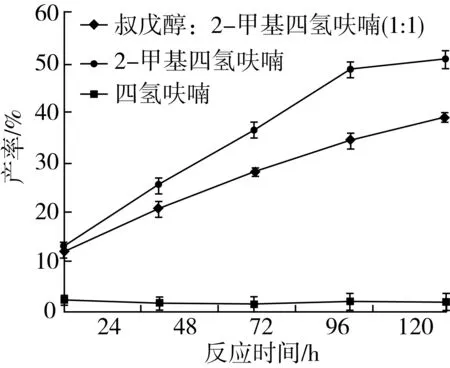
注:反应条件为肉桂酸乙酯0.5 mmol及植物甾醇0.5 mmol,Novozym435脂肪酶80 mg,磁力搅拌器转速300 r/min,反应温度60 ℃,反应时间120 h。图8 有机溶剂对转酯反应的影响
由图8可知,在其他条件相同的情况下,有机溶剂的种类的不同产率差异明显(P<0.05)。随着反应时间的增加,肉桂酸植物甾醇酯的产率均逐渐增大,其中四氢呋喃作为溶剂的反应体系效果较差,原因是四氢呋喃的极性较强,脂肪酶在其中的稳定性较差,很快失活,导致转酯反应无法继续进行。2-甲基四氢呋喃作为溶剂的反应体系中,产率最高,脂肪酶有良好的催化活力,且底物具有较好的溶解性,更有利于其扩散通过酶必需水层进入活性中心。因此,确定最佳反应溶剂为2-甲基四氢呋喃。
2.2.4 脂肪酶添加量对转酯反应的影响
以肉桂酸植物甾醇酯的产率、反应初速度为指标,考察不同脂肪酶添加量对转酯反应的影响,结果如图9所示。
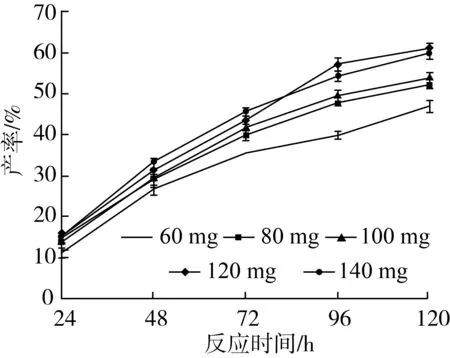
注:反应条件为2-甲基四氢呋喃5 mL,肉桂酸乙酯0.5 mmol及植物甾醇0.5 mmol,Novozym435脂肪酶,磁力搅拌器转速300 r/min,反应温度60 ℃,反应时间120 h。图9 脂肪酶添加量对转酯反应的影响
由图9可知,反应开始时随着酶量的增加,肉桂酸甾醇酯的产率明显增大(P<0.05)。其原因主要是随着酶量的增加,反应速度逐渐提高,参加反应底物的量增多,提高了肉桂酸植物甾醇酯的产率。当反应所加酶量达到120 mg时,产率达到最大,产率可达60.63%。但之后随着酶量继续增加,产率逐渐呈现下降趋势,造成这种结果的原因可能是酶的表面有一层水化层,加入过多的酶就会向体系中引入过多的水,从而使体系的初始水活度提高,使肉桂酸乙酯的水解反应增强,促进了可逆反应进行,使反应肉桂酸植物甾醇酯含量减少,降低了反应的产率。因此,最佳脂肪酶的加入量应为120 mg。
2.2.5 反应温度对转酯反应的影响
由图10可知,反应开始时随着温度的增加,肉桂酸甾醇酯的产率显著增加。温度为55 ℃时,反应初期产率变化较大,说明温度升高加快反应初速度;但温度继续升高,曲线变化平缓,原因是高温提高反应速度的同时,也会加快酶的失活。而且反应长时间处于过高温中,有机溶剂会部分蒸发,反应体系粘度增大,不利于反应介质的传质,而且使得酶活性下降,导致转酯化反应进行缓慢。由图10也可以看出,当温度达到65 ℃时,产率在120 h有所降低,这个结果与Zheng等[19]选择分析讨论结果一致。
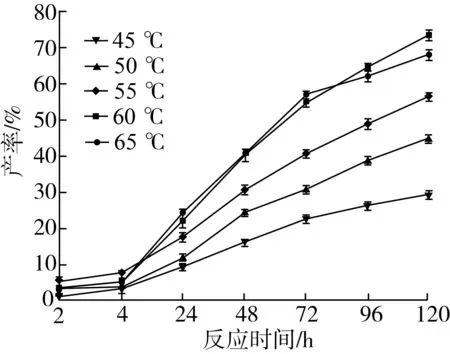
注:反应条件为2-甲基四氢呋喃5 mL,肉桂酸乙酯0.5 mmol及植物甾醇0.5 mmol,Novozym435脂肪酶120 mg,磁力搅拌器转速300 r/min,反应时间120 h。图10 反应温度比对转酯反应的影响
2.2.6 底物比对转酯反应的影响
由图11可知,在转酯反应中,随着反应时间的增加,48 h前底物比对产率影响较小,肉桂酸乙酯、植物甾醇底物比为1∶1时肉桂酸植物甾醇酯产率最高,120 h时产率达到76.34%。将比例升高或减少都导致产率下降。由此可见,植物甾醇浓度过大会影响与肉桂酸乙酯的转酯反应,导致产率下降,说明可能存在酰基供体的底物抑制作用,并且由于植物甾醇量的溶解性较差,对于其浓度的控制有利于后续产物的分离。因此,肉桂酸乙酯与植物甾醇比例为1∶1为最适摩尔比,此结论与Oyeyinka[20]等研究大豆甾醇与乙酸酐转酯反应底物比选择的结果一致。优化后产率可达76.34%。植物甾醇与肉桂酸乙酯的转酯反应同其他方法合成的植物甾醇酯,如植物甾醇醇山梨醇琥珀酸二酯[21]、谷甾醇烟酸酯[22]、甾醇高油酸葵花籽油酯[23]相比,合成的产率相近或较高,并具有成本较低,工艺简单,催化反应条件较温和等优点。
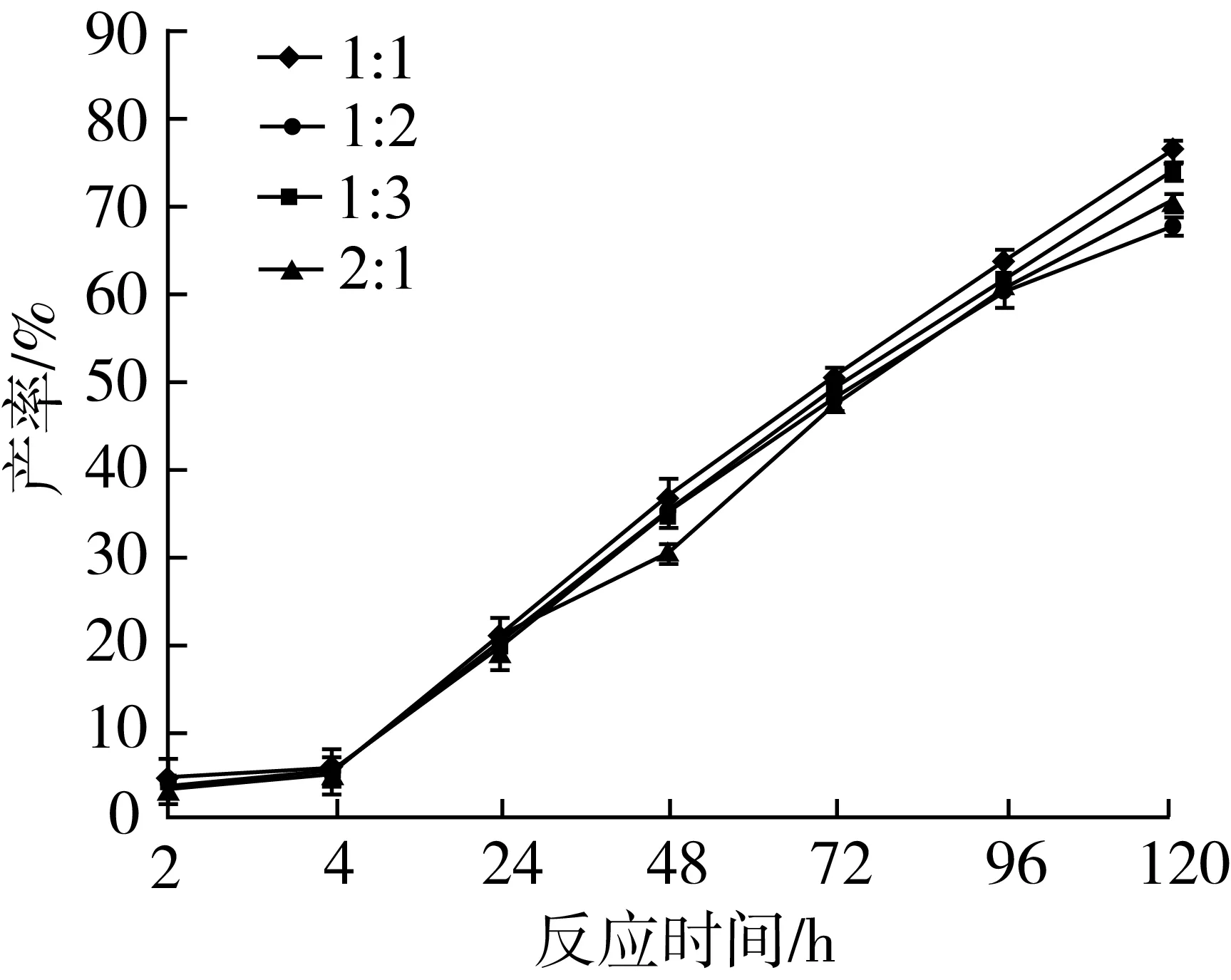
注:反应条件为2-甲基四氢呋喃5 mL,Novozym435脂肪酶120 mg,磁力搅拌器转速300 r/min,反应温度60 ℃,反应时间120 h。图11 底物比对转酯反应的影响
2.3 肉桂酸植物甾醇酯对DPPH·的清除作用
相同条件下,对各浓度梯度的抗坏血酸、茶多酚、肉桂酸乙酯、植物甾醇和肉桂酸植物甾醇酯进行清除DPPH·的能力的测定,结果如图12所示。
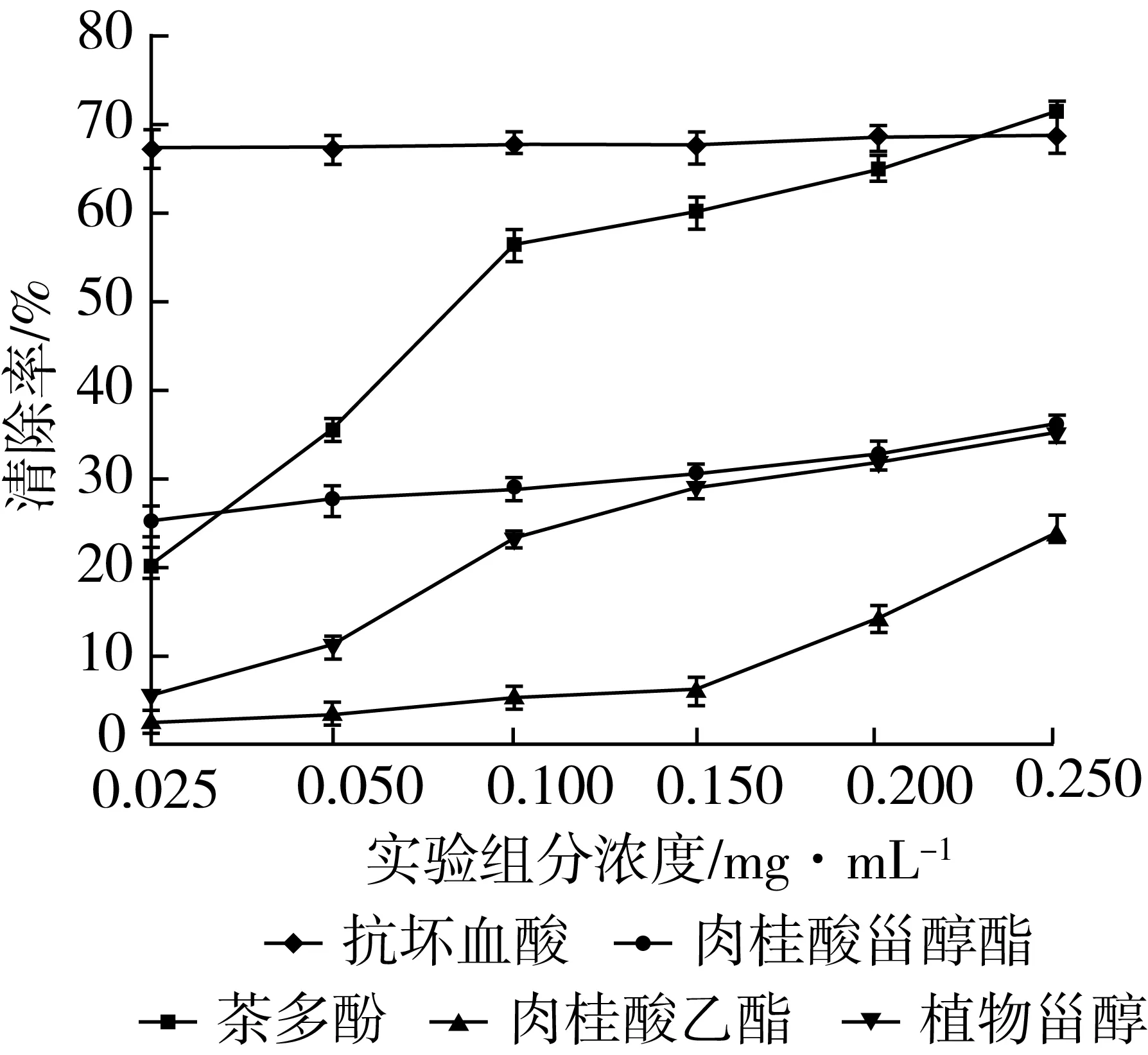
图12 各浓度样品清除DPPH·能力的变化曲线
由图12可知,抗坏血酸在浓度0.025~0.25 mg/mL的范围内,对DPPH·的清除率最大达到60%以上,但随浓度的变化趋势较为平缓;茶多酚随着浓度的上升,清除率的先显著增加,在0.25 mg/mL时大于抗坏血酸,抑制率为70.41%;本实验底物肉桂酸乙酯和植物甾醇相较于同浓度的抗坏血酸和茶多酚对DPPH·清除能力较弱,但是二者均呈上升趋势,肉桂酸乙酯在0.15 mg/mL以后清除率上升明显,而植物甾醇的抑制率开始变缓,并且植物甾醇清除自由基能力稍强于肉桂酸乙酯;此次实验合成的肉桂酸植物甾醇酯,经过测定,具有一定的清除DPPH·的能力,即具有一定的抗氧化性,与底物肉桂酸乙酯和植物甾醇相比,清除自由基的能力稍强,并且改善了植物甾醇的溶解性和稳定性。

表1 各样品清除DPPH·的IC50值
通过表1中各实验样品的IC50值可以看出,所测试的样品各自清除DPPH·的能力大小,IC50值从小到大可表示为:抗坏血酸<茶多酚<肉桂酸甾醇酯<植物甾醇<肉桂酸乙酯。可以看出抗坏血酸红和茶多酚的IC50值较小,清除能力较强,产物肉桂酸植物甾醇酯的IC50值同两个底物肉桂酸乙酯和植物甾醇相比,清除DPPH·效果有所改善。
3 结论
本实验研究了Novozym 435脂肪酶在非水相体系中催化肉桂酸乙酯与植物甾醇合成肉桂酸植物甾醇酯,通过色谱法进行分析确定在最优条件下,酶促合成肉桂酸植物甾醇酯的产率可达76.34%,说明转酯反应的底物转化率较高。通过对产物清除DPPH·能力的测定,发现与植物甾醇相比,具有良好的抗氧化活性。
