新型抗生素葡糖脱乙酰基酶LpxC抑制剂研究进展
2019-09-02王明华张国宁王菊仙王玉成
王明华 张国宁 王菊仙 王玉成
(中国医学科学院 北京协和医学院 医药生物技术研究所,北京 100050)
细菌耐药性尤其是革兰阴性菌的耐药性已成为人类健康的重大威胁。抗生素耐药性每年造成70万人的死亡,如果这一问题无法解决的话,专家们预测,到2050年每年的死亡人数将会达到1000万人[1]。2017年2月,世界卫生组织根据对新型抗生素的迫切需求程度将其分为极为重要、十分重要和中等重要3个类别[2]。列为极为重要的包括耐碳青霉烯类药物的鲍曼不动杆菌(CRA)、铜绿假单胞菌和产超广谱β-内酰胺酶(ESBLs)肠杆菌科(CRE)3种细菌,均为多重耐药性革兰阴性菌。目前临床上极度缺乏安全有效的治疗多重耐药革兰阴性菌感染的药物,全球范围内处于临床研究的候选药物更是寥寥无几。因此迫切需要研发新作用机制的新型抗革兰阴性菌药物。
革兰阴性菌具有一层独特的外膜(OM)结构,它是天然的渗透屏障,能够阻碍药物渗透进入细菌,并激活外排泵,这是导致多种抗生素对革兰阴性菌疗效较差的重要原因。其中类脂A(1ipid A)扮演了一个重要角色,负责脂多糖(LPS)的正确装配以及锚定,同时也是保护细菌抵御外部因子(如抗生素和去污剂)的重要构成部分[3],又是强有力的内毒素,能引发宿主非常强烈甚至致死性的免疫反应(败血性休克),是革兰阴性菌感染致病的重要原因[4-5]。研究表明,LPS对于大部分革兰阴性菌的存活不可或缺,可能关乎LPS在膜蛋白的正确折叠过程。因此抑制其生物合成对细菌来说是致命的。类脂A的生物合成共有10步反应,细胞质中的9种特异性酶参与其中,这9种酶都有成为靶点的可能。由于LpxA催化的第一步反应是可逆反应,平衡常数不理想(只有0.001左右)。因此,LpxC催化的第二步去乙酰化反应成为Lipid A合成的第一个限速步骤,决定着整个合成的效率[6]。所以,通过抑制细菌Lipid A的生物合成来对抗细菌,LpxC便成为了研究人员心目中最为理想的靶点。
1 LpxC简介
LpxC在革兰阴性菌中具有较高的同源性,但与哺乳动物(包括人)的各种酶都没有共同序列[7]。LpxC是控制革兰阴性菌感染的的理想靶标,其特异性抑制剂具有低脱靶和低毒性的特点。由于LpxC是抗革兰阴性菌的全新靶标,不存在已有的耐药性,所以其抑制剂对于临床上耐药革兰阴性菌同样有效。
1.1 LpxC的结构
LpxC是一种锌离子依赖的金属酶,本文以铜绿假单胞菌的LpxC酶为例(图1),其含有两个结构域(domain I和domain II),每个结构域由5个β-折叠结构和两个α-螺旋结构组成,β-折叠包夹着α-螺旋,形成了经典的“β-α-α-β”的三明治结构。这两个结构域虽然存在些许差别(氨基酸序列),但仍具有相同的二维拓扑结构。整个酶的活性部分则位于两个结构域的连接交界处。此外,每个结构域各含有一个β-折叠结构不同的插入区(Insert I和Insert II),插入区I界定了活性位置的范围,由3个β-折叠构成;插入区II则形成一个β-α-β结构式的窄长封闭疏水通道,可与一些疏水性氨基酸残基相连。因此,其可以被疏水长链(如肉豆蔻酸或抑制剂的疏水长链基团)占据,这样可以增加抑制剂与LpxC酶的识别能力,使LpxC活性中心的锌离子与抑制剂的螯合基团(如异羟肟酸等)有效的配位结合。
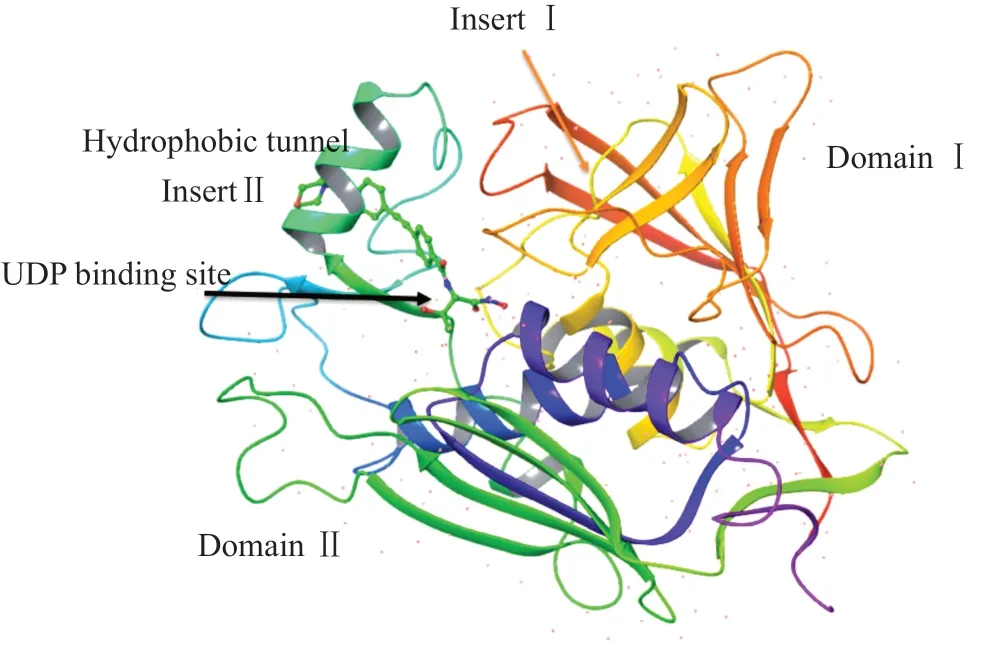
图1 LpxC结构(PDB 5U39)Fig.1 The structure of LpxC (PDB 5U39)
1.2 LpxC的催化机理
LpxC是一种去乙酰化酶,其活性依赖于锌离子,目前得到普遍认可的催化机理主要来自McClerren课题组[8]和Hernick课题组[9]对大肠埃希菌和超嗜热菌中LpxC蛋白的研究。他们提出的催化机理都是基于共轭酸碱理论,但又对组氨酸和苏氨酸在活性区域的作用存在争议。在去乙酰化反应中,McClerren等[8]认为E78作为催化碱夺取锌离子束缚的水分子中的一个质子,促使活化的水分子中的氧原子与底物中乙酰胺基的羰基碳发生亲核反应,形成中间态佯盐,最后葡萄糖胺的末端胺再从E78夺取一个质子,释放出乙酸变成葡萄糖胺。其中,H265(253 of AaLpxC)没有提供也没有夺取质子,仅仅起到稳定佯盐中间体的作用(图2A)。但Hernick等[9]则认为是葡萄糖胺的末端胺是从H265获得质子,而不是E78,而起到稳定佯盐中间体作用的不是H265,而是T191(图2B)。
此外,也有研究报道[10]称磷酸基参与锌离子的催化作用,也有量子力学模型研究表明异羟肟酸类与锌离子形成五配位体中间态,具有很强的结合力[11]。
1.3 LpxC的蛋白调控FtSH
Kdo2-lipidA的合成效率受到膜蛋白降解酶FtsH的调控。当细胞内Kdo2-lipidA过量时,FtsH通过降解关键酶LpxC和KdtA来降低其合成速率[12]。因此通过调控FtSH来降解LpxC酶,从而抑制类脂A的生物合成,也是可以考虑的一个研究策略。
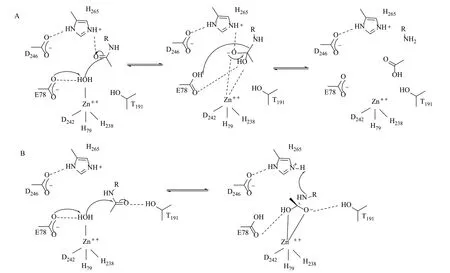
图2 LpxC的催化机理[9]Fig.2 The proposed catalytic mechanisms for LpxC[9]
2 LpxC抑制剂研究进展
自90年代至今的20多年里,LpxC已然成为抗革兰阴性菌药物研究领域非常具有吸引力的靶点之一,针对这一靶点也设计合成了许多类型LpxC抑制剂,所以开发新型LpxC抑制剂也受到广大科研工作者们及制药企业的青睐。到目前为止,LpxC抑制剂按其结构特点可分为以下6大类。
2.1 异羟肟酸类LpxC抑制剂
2.1.1 芳唑啉类
1996年,Onishi等[13]在《科学》杂志上首次报道了芳唑啉类LpxC抑制剂—L-573,655,并合成许多结构相似的衍生物(图3)。其中,化合物L-161,240对大肠埃希菌LpxC酶的Ki值为50nmol/L,而对野生型(envA+)和敏感型大肠埃希菌(envA1)的MIC分别为1~3ng/mL和8~16ng/mL,与氨苄西林相当,优于利福平和红霉素。但其抗菌谱较窄,对铜绿假单胞菌和黏质沙雷菌没有明显抑制活性。
为了便于分析讨论,本文以L-573,655为例,将其分为3部分:芳环片段、杂环片段和螯合基团(图3)。1999年Chen等[14]以L-573,655为先导化合物,设计合成一系列衍生物,酶活性结果表明,螯合基团羟肟酸活性最优,芳环片段连有疏水基团-供电子基(3,4,5-三取代时)活性增加,而亲水或杂环基团对活性不利,这与透膜性有一定关系。2002年Kline等[15]为扩大此类化合物的抗菌谱,以L-161,240为先导化合物,设计合成一系列杂环片段为噁唑啉、噁嗪类、噻唑啉类衍生物,其对铜绿假单胞菌活性最好的达到100nmol/L,其中吸电子基-三氟甲氧基对活性贡献较大。同年,Pirrung等[16]将L-159,692唑啉环替换成噁唑啉环,活性都不如先导化合物;2003年他们首次利用高通量固相合成法设计合成74个苯唑啉类化合物,虽然对铜绿假单胞菌没有活性,但大部分化合物对野生型和敏感性大肠埃希菌有效[17],如化合物5。
2.1.2 底物类
2000年,Jackman等[18]以细菌LpxC的天然底物为先导化合物,设计合成了一类以四氢吡喃环为骨架的LpxC抑制剂,代表化合物为TU-514和TU-517(图4)。这俩化合物对EcLpxC[Ki分别为(650±280)nmol/L和(190±50)nmol/L]和AaLpxC的活性达到较低的微摩尔水平。然而,TU-514可能由于其透膜性较差并未表现出很好的抗菌活性;除此之外,烷基链缩短,对EcLpxC和AaLpxC活性显著下降。
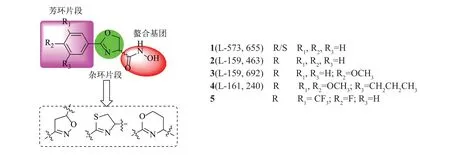
图3 苯唑啉类化合物结构Fig.3 Structures of aryloxazolines

图4 底物类似物结构Fig.4 Structures of substrate analogs
2005年,Li等[19]对TU-514的疏水区以及吡喃环上的羟基进行修饰,遗憾的是,衍生物对E.coliLpxC的抑制率都不尽如意。对吡喃环上的羟基甲醚化或烷基链延长或缩短均使活性下降,而肉豆蔻酰基替换成苯甲酰基则活性保持(化合物14,抑制率为53%)。
2.1.3 磺酰胺类
2002年,Clements等[20]通过金属酶抑制剂库筛选得到了磺酰胺-羟肟酸衍生物,其代表化合物为BB-78484和BB-78485(图5)。这些化合物均含有两个疏水基团,具有非常独特的结构。针对大肠埃希菌LpxC酶,BB-78484和BB-78485的Ki值分别为50和20nmol/L。这俩化合物对另一抗菌靶酶肽脱甲酰基酶(PDF)无明显抑制作用。经体外抗菌活性评价,BB-78484和BB-78485具有广谱抑菌作用(表1)。但随后几乎就没有有关此类化合物的研究报道了。
2.1.4N-芳基-L-苏氨酸类
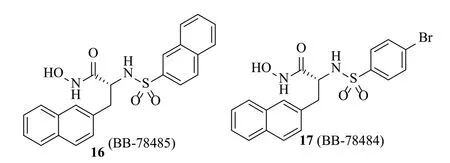
图5 磺酰胺类化合物结构[20]Fig.5 Structures of sulfonamides[20]
2004年,Anderson等[21]报道了一种新型的N-芳基-L-苏氨酸异羟肟酸类LpxC抑制剂,其中代表性的化合物是CHIR-090(图6),由McClerrend等[8]开发。CHIR-090能同时抑制大肠埃希菌(MIC:0.20μg/mL)和铜绿假单胞菌(MIC:1.6μg/mL)的生长,且与妥布霉素和环丙沙星的抗菌活性相当,但是对豌豆根瘤菌(Rhizhobium leguminosarum)的抑制活性(Ki为340nmol/L)不高。随后研究者们通过构建解析不同LpxC酶与CHIR-090的共晶复合物,开启了LpxC抑制剂对革兰性阴性菌的的抗菌谱研究,以及基于CHIR-090的结构修饰和优化[22-23]。
目前绝大多数活性较高的化合物都是通过对CHIR-090结构优化和骨架跃迁得到的。为了便于分析讨论,将CHIR-090结构拆分成5部分(图6):P1为锌离子螯合基团;P2区不同取代基对抗菌活性有不同的影响;P3为连接疏水基团和螯合基团的连接子;P4区为占据LpxC酶疏水口袋的疏水片段;P5为末端基团(溶剂暴露区),一般对抗菌活性影响不大,主要改善分子的理化或代谢性质。

表1 磺酰胺类LpxC对不同致病菌的活性[20]Tab.1 Activities of sulfonamides against a range of pathogens[20]
2011年,Lee等[24-27]报道了以丁二炔为疏水骨架的化合物LPC-009(MIC:E.coli0.05μg/mL;P.aeruginosa0.74μg/mL);为解决溶解度问题,又在末端引入氨基得到LPC-011(MIC:E.coli0.03μg/mL;P.aeruginosa0.5μg/mL),抑菌活性都要明显优于CHIR-090。随后他们向P2区引入了一些芳(杂)环等,活性均有所降低。2016年又报道[28]了一个具有广谱抗菌活性的化合物LPC-058,其对大肠埃希菌LpxC的Ki值达到了3.5pmol/L,对大肠埃希菌、铜绿假单胞菌、对鲍曼不动杆菌的MIC分别为0.0018、0.167和0.39μg/mL,这也是少有的对鲍曼不动杆菌有活性的化合物。
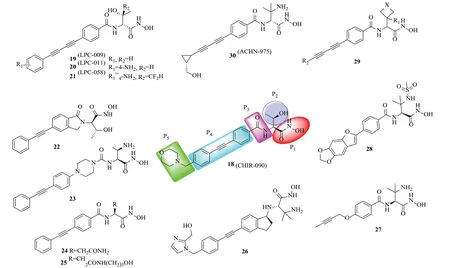
图6 N-芳基-L-苏氨酸类化合物结构Fig.6 Structures of N-aroyl-L-threonines
2011年,Mansoor等[29]将P3区替换为苯并内酰胺和脲类衍生物(如化合物22~23);苯并内酰胺类对LpxC酶活性弱于开环的,而部分脲类具有广谱抗菌活性。2013年,Hale等[30]将P2区替换为不同取代氨基酸(谷氨酸、环氨基酸等),其中含谷氨酸片段活性较好,化合物24对铜绿假单胞菌IC50为4.4nmol/L, 化合物25对大肠埃希菌IC50为1.3nmol/L。2015年,杨玉社课题组[31]在P5区引入曲酸及甲基砜片段,其中引入曲酸片段使活性丢失,部分甲基砜类化合物活性与CHIR-090相当,在肝微粒体中的代谢性质要优于对照。2018年,他们又报道[32]P1区羟胺活性优于邻苯二胺;P2区2-氨基异丙基优于2-羟基乙基,且代谢更稳定;P3区磺酰基无协同作用,使活性降低;P4区将苯环换成取代苯环或噁唑烷酮环或吡啶都会不同程度上降低活性。2017年,Jing等[33]报道P3区为二环类似物,代表化合物26(MIC:E.coli0.5μg/mL;P.aeruginosa1μg/mL),但是其具有静脉急性毒性并抑制钠离子通道site II(97%,25μmol/L,IC50=400nmol/L)。 诺华公司Piizzi等[34]为改善水溶性和降低毒性,对P2区进行了衍生-引入羧基及哌嗪酮等,活性均有所提高;将P4区二苯乙炔基替换为不同取代苯,其中炔丙氧基最优,如化合物27,对野生铜绿假单胞菌IC50为1.5nmol/L,MIC为1.0μg/mL,MDR铜绿假单胞菌MIC90为2μg/mL。Kawai等[35]报道了P2区为甲磺酰胺类化合物,主要探讨了疏水区结构对活性的影响,其中化合物28活性最优(E.coliMIC=0.063μg/mL,K.pneumoniaeMIC=0.5μg/mL和P.aeruginosaMIC=0.5μg/mL)。Achaogen公司在一项专利中又报道[36]了一类抗菌活性较优的化合物,将P2区变为四、五元饱和杂环,如化合物29。
该类抑制剂中研究最为前沿的是Achaogen公司开发的ACHN-975(铜绿假单胞菌MIC:0.25μg/mL),该化合物曾进入I期临床研究,它无论对于敏感型还是耐药型铜绿假单胞菌的抗菌活性都明显优于临床上常用的妥布霉素、环丙沙星、头孢他啶、亚胺培南和黏菌素等临床上常用的一、二线抗菌药物[37]。然而该化合物由于局部注射耐受性问题从临床试验撤回。ACHN-975的临床前和临床研究表明,N-芳基-L-苏氨酸类是一类具有广阔开发前景的抗革兰阴性菌化合物。
2.1.5 烷基砜类
2012年,辉瑞全球研发中心Brown等[38]报道了烷基砜-异羟肟酸类抑制剂,代表化合物为1a(图7)。化合物1a对铜绿假单胞菌LpxC酶的抑制活性达到纳摩尔级水平(IC50为1.37nmol/L),与阳性对照CHIR-090(IC50为2.05nmol/L)相当,但其对铜绿假单胞菌(PAO1)的抑菌活性比CHIR-090要好(MIC分别为0.125和1μg/mL)。此后,Montgomery等[39]对先导物1a进行了相应的结构修饰和改造,得到系列吡啶酮烷基砜类化合物(如2a和33),此类化合物中大部分都与先导物1a的抑酶抗菌活性相当。其中2a对铜绿假单胞菌LpxC酶的IC50为3.6nmol/L,对铜绿假单胞菌(PAO1)的MIC为0.5μg/mL。McAllister等[40]对疏水区进行了SAR探索,发现5-三唑类(化合物34)和异噁唑类(化合物35)对铜绿假单胞菌有一定活性,MIC均为0.5μg/mL。Abdel-Magid等[41]申请的专利中报道了一类吡咯[1,2-C]咪唑-3-酮类化合物36,其对大肠埃希菌和铜绿假单胞菌的MIC分别为≤0.063和0.5μg/mL。同年诺华公司的专利[42]中报道一异噁唑类化合物37,对大肠埃希菌和铜绿假单胞菌的MIC分别为0.25和2μg/mL;葛兰素史克公司也报道[43]了一类苯并嗪类化合物38,对大肠埃希菌和铜绿假单胞菌的MIC分别为≤0.03和0.06μg/mL。
此类抑制剂的研究者主要为制药企业,也是一类研究较多,具有广阔开发前景的抗革兰阴性菌化合物。
2.1.6 呋喃乙酸及苄氧乙酸类
2012年,Oddo等[44]合成了一类含糖苷类的LpxC抑制剂,利用拼合原理和构象限制策略,用C-糖苷骨架替换苏氨酸酰胺片段(化合物39,图8)。2013年,Jana等[45]对上述化合物疏水片段进行修饰得到C-三氮唑-α-D-呋喃糖苷类衍生物(化合物40),利用琼脂扩散法表明其无抗菌活性及酶抑制活性,可能是空间构象限制不利于与酶的结合;2013年[46]合成了C-三氮唑-β-D-呋喃糖苷类,同样也没有活性;随后又设计了C-乙炔基呋喃糖甘类(化合物41)[47],活性也不理想。2013年,Löppenberg等[48]也报道了将化合物39疏水片段替换为不同长度、构型、柔性的亲脂性链。活性结果表明,相对较短、弯曲和柔性的亲酯链没有抗大肠埃希菌活性,而较长刚性线性如二苯基丁二炔活性较好,对大肠埃希菌Ki值为4.4μmol/L,其中糖苷开环产物(化合物42)活性优于构象限制的。随后Szermerski等[49]和Tangherlini等[50]基于化合物42,通过去掉其中一个羟甲基,设计了苄氧基乙异羟肟酸类,但活性也不理想。
2.1.7 四氢吡喃类
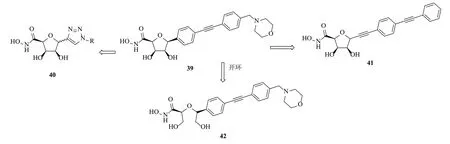
图8 呋喃乙酸及苄氧乙酸类化合物结构Fig.8 Structures of C-glucosides

图9 四氢吡喃类化合物结构Fig.9 Structures of tetrahydropyrans
2014年,阿斯利康Murphy-Benenato等[51]对异羟肟酸化合物库进行筛选,得到化合物43(图9),是基质金属蛋白酶(MMP-2,-9,-8,-13)抑制剂RS-130830的衍生物。其对铜绿假单胞菌LpxC IC50为(7.4±2.6)nmol/L,MIC为50μmol/L,但对野生型大肠埃希菌活性较弱(E.coliARC523 MIC>200μmol/L)。以此化合物为苗头化合物,保留四氢吡喃环对疏水区进行优化,其中化合物44对铜绿假单胞菌IC50为(4.4 ±1.6)nmol/L,细胞水平活性相似,对大肠埃希菌活性提高(MIC:12.5μmol/L)。化合物45~46也呈现出较好活性,但其对MMP(基质金属蛋白酶)的活性并未消除,这就增加了脱靶效应。而将吡喃环变为环己醇,使活性降低。
与西班牙语类似,贯穿全文的卡拉米洛披肩亦是受到外来文化的影响:“它是由印度妇女包裹孩子的布和西班牙披肩上打结的穗结合而成,中国宫廷的绸刺绣出口到马尼拉,通过西班牙帆船进而到阿卡普尔科。殖民时期,由于禁止买西班牙人穿的那种衣服,墨西哥人开始用当地产的织布机织布,一种长长的窄窄的、潜移默化地收到外国影响的围巾。(96)
2.1.8 噁唑烷酮类
2016年,Kurasaki等[52]将噁唑烷酮替换苏氨酸,得到限制构象的噁唑烷酮类化合物,其中化合物48(图10)对大肠埃希菌的IC50为6nmol/L,MIC为0.016μg/mL,具有较低的外排率。
2.1.9 1,2,3-三唑连接核苷-氨基酸类
2017年,Malkowski等[53]依据底物设计了4个三唑连接核苷轭合物(图11),只测定了均衡校正电子相互作用的能量。在真空模型中,化合物51具有较好的相互作用能;通过计算机模拟发现三氮唑也能很好地与锌离子结合,这加强了与锌离子的相互作用,遗憾的是并未测抗革兰阴性菌的活性。
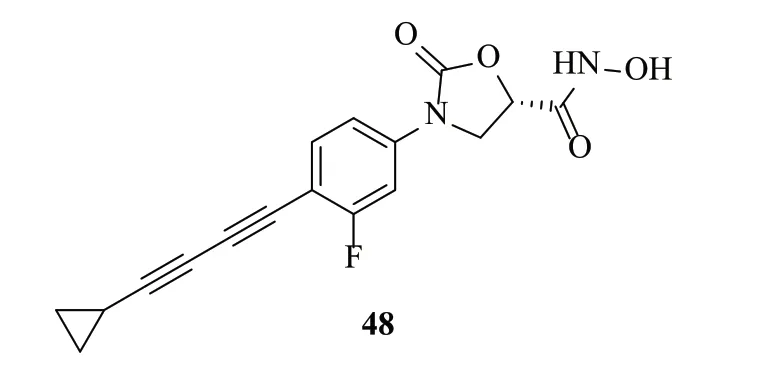
图10 噁唑烷酮类化合物结构Fig.10 Structures of oxazolidinones
2.2 磷酸类LpxC抑制剂
2000年,Jackman等[18]在设计底物类似物时,用磷酸基团代替异羟肟酸基团引入到TU-521中得到了四氢吡喃糖磷酸类抑制剂(化合物53)(图12),此化合物对大肠埃希菌[(115±33)μmol/L]和超嗜热菌LpxC[(192±38)μmol/L] 的抑制活性相对较弱,不如异羟肟酸类。2002年,Pirrung等[16]也设计合成了2个芳唑啉磷酸类LpxC抑制剂(化合物54和55),化合物54对大肠埃希菌LpxC酶[IC50=(4±2)μmol/L]的抑制活性与阳性对照L-159692相当(IC50=3μmol/L)。2016年,王玉成等[54]将磷酸基引入CHIR-090得到化合物56,对大肠埃希菌和铜绿假单胞菌MIC分别为0.28和2.06μg/mL,与CHIR-090相当。
2.3 脂肪族羧酸和两性苯甲酸类LpxC抑制剂
2007年,Shin等[55]报道了一类含有长疏水脂肪烷基链的羧酸或苯甲酸衍生物(图13),此类化合物为两亲性分子,主要靶向窄长的疏水通道。虽然与LpxC酶结合活性Kd值最低达到了0.9μmol/L,但此类化合物的抗菌活性不理想,可能由于羧基的螯合能力较弱。另一方面,疏水脂肪链的长短对与酶的结合活性有较大影响,最佳碳原子数为6~12个;碳原子数为12时活性最佳(Kd值为0.9μmol/L),少于6个时不能与疏水通道发生有效结合。由于此类化合物抗菌活性太弱,没有进一步研究价值。
2.4 乙内酰脲类LpxC抑制剂
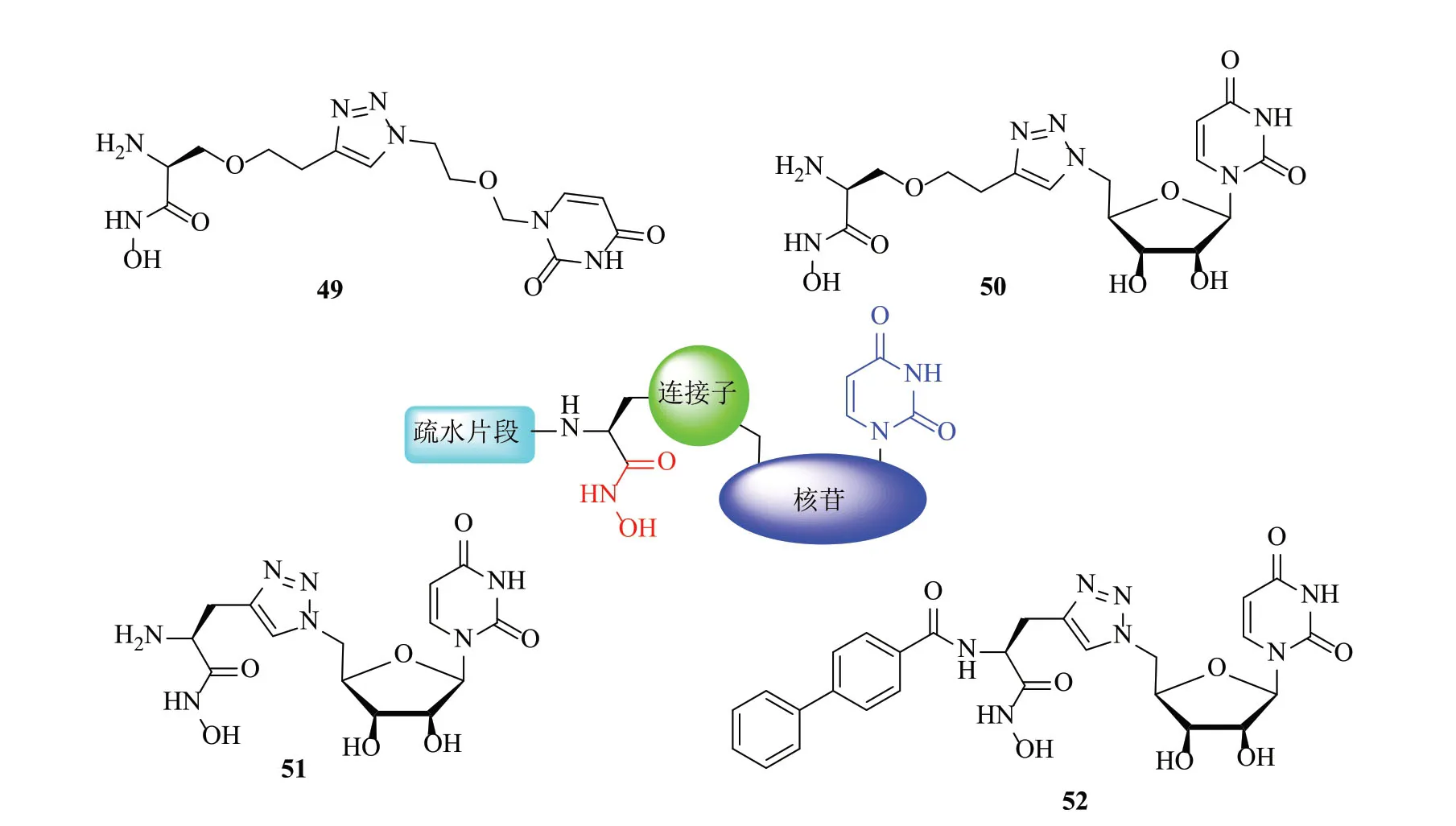
图11 三唑连接核苷-氨基酸类化合物结构[53]Fig.11 Structures of 1,2,3-triazole-linked nucleoside-amino acid conjugates[53]
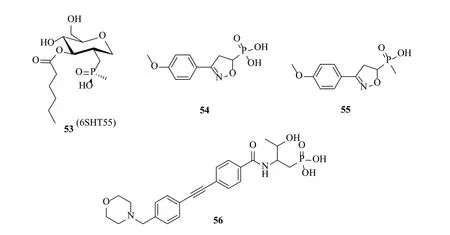
图12 磷酸类化合物结构Fig.12 Structures of phosphates

图13 脂肪族羧酸和两性苯甲酸类化合物结构Fig.13 Structures of aliphatic carboxylic acid and amphipathic benzoic acids
2008年,Schering公司利用电子等排体乙内酰脲代替异羟肟酸,设计合成了一系列螯合基团为乙内酰脲类的化合物[56](图14),期望降低LpxC抑制剂的毒性,提高选择性。其中,化合物59对LpxC抑制活性最优(IC50:0.5μmol/L)。虽然此类化合物具有适度的LpxC酶活性,但其具有较广谱的抗G-菌活性,值得进一步研究。
2.5 尿嘧啶核苷类LpxC抑制剂
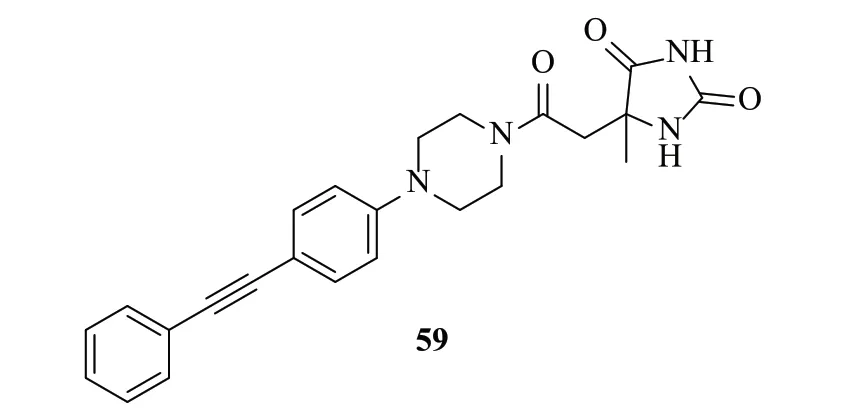
图14 乙内酰脲类化合物结构Fig.14 Structures of hydantoins
当前,对“基于UDP位点”设计合成的LpxC抑制剂的研究甚少,但化合物52和60为我们打开了设计LpxC抑制剂的新思路—如何利用UDP位点设计出选择性更高、结合性更好的LpxC抑制剂,在现有活性较好的抑制剂的结构基础上引入与UDP口袋有效结合的基团不失为一个研究策略,但也充满艰难,因为其对抗菌活性影响很大。
2.6 其他类
Forge Therapeutics公司报道了一类非异羟肟酸类LpxC抑制剂,其中FG-944表现出极好体内外抗菌活性,其对大肠埃希菌MIC为0.25μg/mL,肺炎克雷伯菌1μg/mL,对多重耐药菌株均有同等效果,并且与其他锌离子金属蛋白没有交叉相互作用(ACE1、HDACs和CAII),遗憾的是公司并没有披露相关结构,但本文根据其专利[58]推测,其结构可能为羟基吡啶酮和羟基嘧啶酮类化合物(图16)。
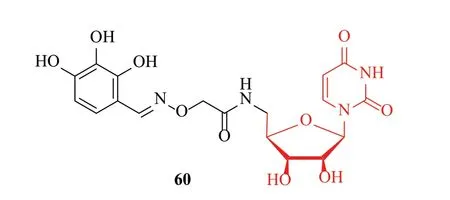
图15 尿嘧啶核苷类化合物结构Fig.15 Structures of uridines
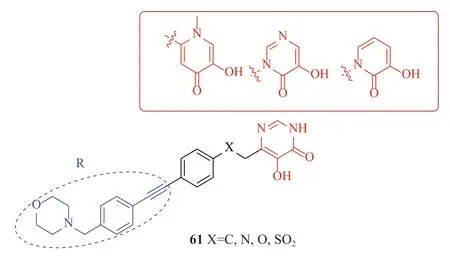
图16 羟基吡啶酮和羟基嘧啶酮类化合物结构Fig.16 Structures of hydroxypyridinones and hydroxypyrimidinones
3 结语与展望
随着多重耐药革兰阴性菌的日益严重,迫切需要开发具有新的作用机制的新型抗生素来对抗这些病原体。LpxC在几乎所有革兰阴性菌种中具有高度序列保守性,并且与任何哺乳动物蛋白质缺乏序列同源性,使得LpxC成为极具吸引力的药物靶标。目前,虽然市场上还没有批准的LpxC抑制剂,但在过去20多年,学术界和制药业都在努力开发有效的LpxC抑制剂。其中,N-芳酰基-L-苏氨酸类和烷基砜类LpxC抑制剂是最有希望的类别。
然而,尽管许多报道的化合物具有很好的临床前数据,但除了最近撤回的ACHN-975之外,目前还没有其他LpxC抑制剂进入临床试验。因此,我们推测LpxC抑制剂可能存在如下缺点:(1)疏水腔活性必需的亲脂性片段不仅可能增加由于非特异性亲脂相互作用引起的脱靶效应,而且还可能导致较差的物理化学性质,例如溶解度低、蛋白结合率高等;(2)锌离子螯合基团-异羟肟酸在药动学性质方面是不利的,它可以通过轭合快速代谢,释放出有毒的羟胺;还可能引起与不同的人锌离子依赖性蛋白的非特异性结合,如MMP、HDAC(组蛋白脱乙酰酶)、碳酸酐酶等,从而引起不可预测的副作用;(3)一些致病菌如鲍曼不动杆菌、脑膜炎奈瑟菌和黏膜炎莫拉菌可以在没有脂质A的情况下存活,而LpxC抑制剂缺乏针对此类菌种的活性;(4)单一针对LpxC酶,很容易产生耐药性。
因此,此类研究应该寻找新的锌离子螯合基团取代异羟肟酸部分,引入能深入渗透UDP结合位点的取代基(缩短抑制剂的亲脂侧链来补偿活性的降低),以期改善LpxC抑制剂的抗菌活性、选择性和理化性质,并降低毒性。
