肝硬化血小板减少的病理生理发生机制
2019-07-26肖函王利
肖函,王利
作者单位:400010重庆市 重庆医科大学附属第一医院血液科
血小板减少是肝硬化并发的最常见血液系统病症。以往认为肝硬化患者血小板减少主要系门静脉高压脾脏长大、脾脏功能亢进,脾脏巨噬细胞吞噬及脾脏滞留大量血小板所致。近年新认识的肝硬化血小板减少病理生理机制表明,除了脾脏因素外,其它导致血小板减少的机制应该引起临床医师的关注,了解这些新的理念,有助于调整临床治疗策略。
1 血小板生成的生理过程
巨核细胞是从骨髓造血干细胞、原始巨核细胞、幼稚巨核细胞分化而来的细胞,是正常骨髓中一种能生成血小板的成熟细胞,核很大,数量非常少。成熟的巨核细胞又经历了:①颗粒型巨核细胞:胞核大,胞质丰富,染粉红色,夹杂有蓝色,质内含有大量细小的紫红色颗粒,常聚集成簇,但无血小板形成;②产生血小板型巨核细胞(产板细胞):胞体巨大,有时可达100μm,胞核高度分叶状,胞质呈均匀粉红色,质内充满大小不等的紫红色颗粒或血小板。胞膜不清晰,多呈伪足状,其内侧及外侧常有血小板的堆集;③裸核型巨核细胞:产板细胞的胞浆解体后,释放出大量血小板,仅剩一裸核。这些被细胞膜包围的与巨核细胞胞质分离的小块胞质,进入血循环成为血小板。每个巨核细胞产生约300~4000个血小板。新生血小板先通过脾脏,约有1/3在此贮存,约2/3在末梢血循环中。贮存的血小板可与进入循环中的血小板自由交换,以维持血中的正常量。血小板寿命约7~14天,每天约更新总量的1/10,衰老的血小板大多在脾脏中被清除。循环中血小板正常值为100~300×109/L。
血小板的生成受血液中的血小板生成素(thrombopoietin,TPO)调节。TPO是一种糖蛋白激素,由位于染色体3长臂q26.3-27的基因编码,包含两个结构域:氨基末端和羧基末端。氨基末端与促红细胞生成素同源。核糖核酸印迹分析表明,人TPO信使核糖核酸在成人肝脏及肾脏都有表达。TPO基因敲除的动物模型研究发现,外周循环60%TPO主要由肝细胞产生,少量来自于肾脏、骨髓等其他器官[1]。来自肝脏的TPO通过恒定的速率释放到外周循环,不受外周血小板数目的影响[2]。
TPO在血小板的生成及促进血小板聚集的生理功能中起着重要作用。TPO通过与不同细胞表面的血小板生成素受体(thrombopoietin receptor,TPOR)结合,发挥不同的生理作用。TPOR主要位于巨核细胞和血小板表面,也分布于一些造血前体细胞,但分布密度更低。当TPO和造血前体细胞表面的TPOR结合时,虽不能直接刺激造血干细胞朝巨系分化,但会增强巨核系前体细胞的存活和增殖,进而增加血小板的产生[3]。当TPO与巨核细胞表面的TPOR结合时,形成激活二聚体 ,进而激活JAK和STAT信号通路,从而促进巨核细胞生长和成熟,增加血小板的产生(图1)[4]。
当TPO与血小板表面的TPOR结合时,形成的受体-配体复合物被内吞入血小板内,TPO被降解,并使TPOR不再出现在血小板表面[5-7]。TPO的清除主要是通过与血小板表面的TPOR结合,然后被吞噬降解。当血清TPO水平增高后,外周血小板数量就会增多,当外周血小板数量增多后,更多的TPO就被降解,血清TPO水平就下降,外周血小板的数量随之下降,这被称为循环TPO的“海绵效应”,如此负反馈使得血液血小板处于一个比较稳定的水平(图2)。
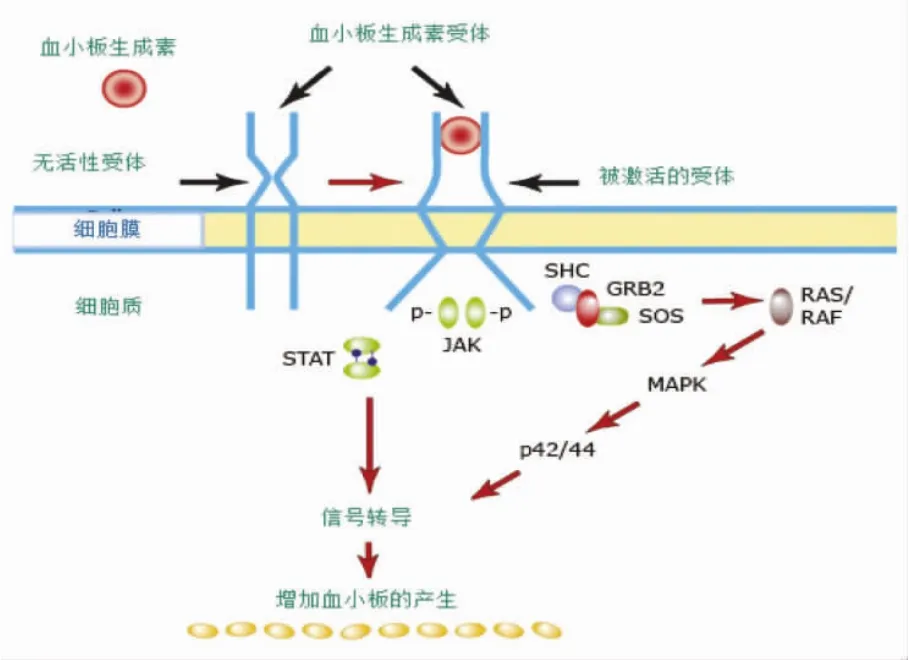
图1 血小板生成素受体激活与血小板产生模拟图(Blood,2007,109:4607.)
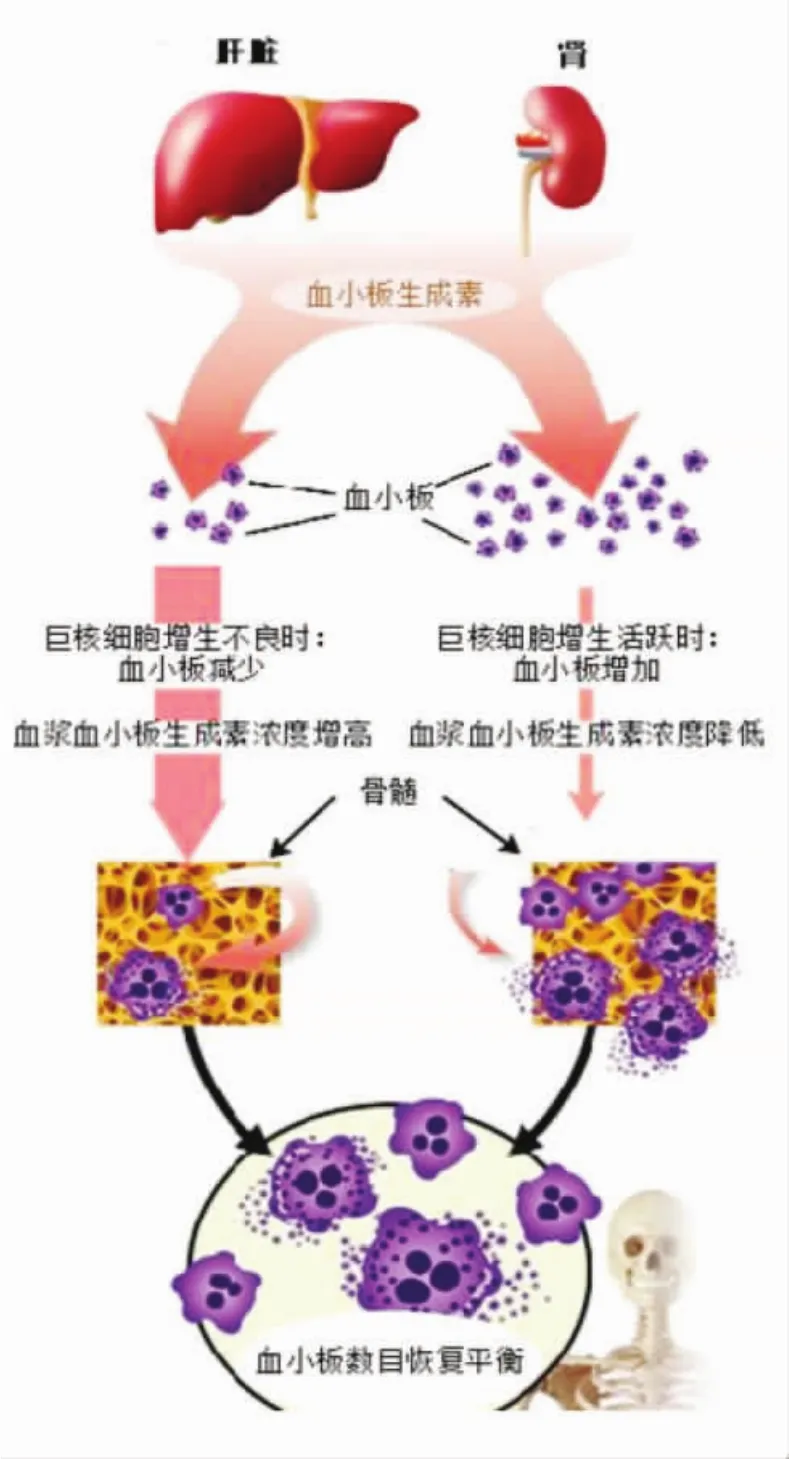
图2 血小板生成的生理调控机制(Liver Int,2017,37:778-793)
当TPO与血小板上的受体结合后,还可以诱导促细胞分裂剂激酶及其他小分子上的酪氨酸残基磷酸化,并激活一系列的信号通路,通过增强二磷酸腺苷和胶原蛋白等血小板激活剂的作用,促进血小板聚集[8,9]。
2 肝硬化对血小板数量的影响
2.1 血小板生成减少 慢性肝病和肝硬化对血小板生成的影响主要涉及以下两方面:1,病毒感染和长期大量饮酒是肝硬化最常见的两大原因,这些病因均可抑制骨髓造血干细胞、原始巨核细胞、幼稚巨核细胞的分化。体外实验观察到,乙型肝炎病毒、丙型肝炎病毒可直接抑制骨髓祖细胞的生长和分化[10-12],血小板产生减少。流行病学研究结果提示,乙型肝炎病毒感染和丙型肝炎病毒核糖核酸阳性是引起血小板减少的独立因素[13-15]。对于普通成年人,长期酒精摄入超过80 g/d,可表现为血小板下降,具体机制不详。对这些饮酒者在戒酒的基础上予以合理饮食,血小板可在1~2周内升至600 000~900 000/毫升,然后在后续的7~10天内恢复正常[16]。
2.2 肝脏TPO合成减少 在骨髓功能受抑,如再生障碍性贫血和化疗后骨髓抑制或肝硬化患者,都会出现血小板减少,前者血浆TPO水平超过正常值的10~20倍[17],而后者TPO却处于一个“正常”或者较低的水平[18-20]。由于两者都有血小板减少,所以外周循环血清TPO水平均下降,但在骨髓衰竭患者,肝细胞仍能产生并释放TPO到外周循环中,故血TPO水平显著上升。在肝硬化患者,由于肝细胞减少,且TPO信使核糖核酸产生较健康人群慢[2],TPO产量下降,血TPO处于一个“正常”或者较低的水平。正常人循环TPO水平为94.7±35.9 pg/ml,肝硬化患者TPO显著降至29.9±18.1 pg/ml。值得注意的是,评判肝脏是否存在TPO产生减少,不应以TPO是否在“正常范围”来判断,而应该结合血小板的实际数量来判断。
2.3 血小板消耗与破坏增加 慢性病毒(特别是丙型肝炎病毒)感染所致肝硬化可产生血小板表面抗原的自身抗体,即病毒某些组分与血小板组分相似,机体免疫系统错误识别,产生自身免疫性T细胞和抗血小板抗体[21]。这些血小板表面抗原分子模拟的自身抗体还可通过增加肝脾内吞噬细胞的作用,加速对血小板的破坏。在自身免疫性血小板减少性紫癜患者中,排除严重肝病患者,约30%患者感染丙型肝炎病毒;反之,丙型肝炎病毒感染人群自身免疫性血小板减少性紫癜发病率高于非丙型肝炎病毒感染人群(HR=1.8,95%CI 1.4~2.3)[22]。此外,肝硬化患者用药不慎,除发生药物性肝损伤外,还可致药物诱导的自身免疫性血小板减少。
2.4 脾功能亢进 肝硬化脾脏长大,脾内巨噬细胞数量增加、Toll样受体-4过表达,使其处于活化状态;脾内microRNA-615-3p高表达提示脾内巨噬细胞吞噬能力增强,增加了对血小板的破坏[23,24]。
2.5 温韦伯氏因子裂解酶(a disintegrin and metalloprotease with a thrombospondin type 1 motif,member 13,ADAMTS13)产生减少 温韦伯氏因子(von Willebrand factor,vWF)是血浆重要成分之一,其与血小板膜上的受体糖蛋白1b(glycoprotein,GP-Ib)结合后,使血小板黏附于血管内皮损伤部位,促进血小板聚集。肝脏星状细胞合成的ADAMTS13是一种类似于剪刀的蛋白,可以将由血管内皮产生的体积较大的vWF串状多聚体降解成体积更小的多聚体,从而将其清除[25]。血管内液体成分和不同颗粒成分的运动速率存在一定的差异,不同成分之间存在着一定的剪切力,这种剪切力一方面促进vWF多聚体的立体结构由球形向长链型改变,进而促进vWF-血小板的聚集;另一方面这种剪切力又促进ADAMST13对vWF多聚体的切开作用。因此,正常情况下,血管内处于一种平衡状态,不会出现血小板大量聚集消耗的情况。失代偿期肝硬化患者循环血ADAMTS13减少[26,27],使得体积较大的vWF长链多聚体难以降解,vWF与血小板的GP1b受体结合,血小板大量粘附积聚在血管内皮上,血小板因此消耗过多而减少,其机制类似于血栓性脉管炎(图3),但血小板消耗程度往往不似弥漫性血管内凝血那样严重[28]。
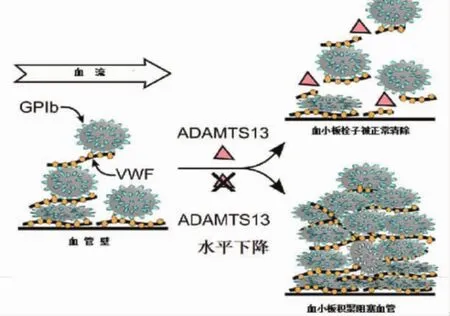
图3 ADAMTS13水平与血小板消耗机制
2.6 循环血小板的分布异常 肝硬化血小板减少还与血小板分布异常有关,主要有:脾脏隔离作用。门静脉高压使得脾脏血液回流受阻,脾脏瘀血肿大,滞留在脾脏血管池内的血小板数量明显增加。在脾脏极度增大的情况下,脾内血小板滞留百分比可由生理状态下的30%增加至90%,外周血血小板数目明显下降。如果仅是单纯脾脏体积增大而肝功能正常时,如 EB病毒(Epstein-Barr virus,EBV)、巨细胞病毒感染后导致的脾肿大,循环中血小板减少主要是分布异常所致,其体内总的血小板数量和生存时间是保持相对正常的,临床上很少会因此发生出血事件[29]。近年研究表明,肝硬化脾肿大并不单纯是瘀血肿大,脾脏白髓部分增殖体积显著高于红髓部分,脾脏纤维化程度增加,脾脏的这种隔离作用对循环血小板减少并非是关键因素。出现血小板卫星现象,在部分患者外周血检查中,可以观察到血小板几乎在每一个多核细胞周围都形成了一圈卫星灶,即血小板卫星现象(图4),此时,血小板计数可明显低于真实水平。在这样的血标本中加入枸橼酸钠,这种卫星现象随即消失,血小板计数恢复正常[30,31]。血小板卫星现象并不太常见,其原因也不甚明确。但了解这种现象的存在,有助于我们恰当地判断患者循环中血小板的真实水平[32]。
肝硬化血小板减少涉及多方面机制,全面地认识这些机制不仅可明智地对待血小板减少,也可增加对早期肝病的诊断。

图 4 血小板卫星现象(N Engl J Med,1998,338:591)
