NLRC4炎性小体及相关疾病
2019-07-16赵梦珠
罗 艺,赵梦珠,吴 迪,沈 敏
固有免疫是机体抵御病原微生物入侵的第一道防线,主要通过模式识别受体(pattern recognition receptors, PRRs)识别病原体相关分子模式(pathogen-associated molecular patterns, PAMPs)或内源性的损伤相关分子模式(damage associated molecular patterns, DAMPs),激活下游信号通路,促进炎症因子的分泌及炎症的产生。核苷酸结合寡聚化结构域(nucleotide-binding oligomerization domain,NOD)样受体(NOD-like receptor, NLR)家族是位于细胞质中的一类PRRs,可划分为多个亚家族,包括NLRA、NLRB、NLRC和NLRP等。NLR识别并结合配体后,可装配形成炎性小体,炎性小体活化后能激活半胱氨酸天冬氨酸特异蛋白酶-1(caspase-1),促进白介素(interleukine,IL)-1β、IL-18前体的切割、成熟和分泌,参与炎症反应、损伤修复等过程[1]。目前研究较广泛的炎性小体主要有NLRP1、NLRP3、NLRC4、NLRP6、NLRP12、黑色素瘤缺乏因子2(absent in melanoma 2,AIM2)炎性小体等。本文主要介绍NLRC4炎性小体的构成、活化机制、作用机制及其相关疾病的研究进展。
1 NLRC4炎性小体的构成
炎性小体由受体蛋白、凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing CARD,ASC)和caspase-1前体(pro-caspase-1)三个部分构成,其中受体蛋白主要包括NLRs(如NLRP1、NLRP3、NLRC4)和HIN200家族蛋白(如AIM2)[2]。
NLRC4炎性小体中的受体蛋白为NLRC4(图1)。NLRC4又被称为IPAF(IL-1β converting enzyme protease activating factor, IL-1β转换酶蛋白激活因子),为NLR家族成员。NLRC4由3个结构域构成,其N端为caspase募集结构域(caspase recruitment domain, CARD),是效应结构域,可募集并活化pro-caspase-1,负责下游信号转导。中间为NOD结构域,又称NACHT结构域,是NLR家族成员共有的特征性结构域,包括核苷酸结合域(nucleotide binding domain, NBD)、2个螺旋结构域(helical domain, HD1、HD2)和翼状螺旋结构域(winged-helical domain, WHD),自N端至C端依次为NBD-HD1-WHD-HD2,可介导NLR分子的寡聚反应,改变其构型。C端为亮氨酸重复结构域(leucin rich repeat, LRRs),负责识别和结合PAMP等配体。ASC为衔接蛋白,由热蛋白结构域(pyrin domain,PYD)和CARD结构域组成,其一端与NLRC4蛋白连接,另一端通过自身的CARD结构域募集pro-caspase-1。Pro-caspase-1为无活性形式,激活后成为caspase-1,是炎性小体的重要组成部分,可介导炎症反应和诱导细胞焦亡(pyroptosis)。
与NLRP3和AIM2依赖于ASC募集pro-caspase-1不同,由于NLRC4存在CARD结构域,NLRC4可在没有ASC的情况下直接募集和活化pro-caspase-1[3]。另有研究表明,若缺乏ASC,则caspase-1的活化和IL-1β、IL-18的分泌均减少,而细胞焦亡过程不受影响,说明ASC在促炎细胞因子的分泌过程中有重要作用,但具体机制尚不明确[4-5]。
2 NLRC4炎性小体的活化和作用机制
在无活化信号时,NLRC4通过LRR结构域折叠使其靠近NOD结构域,抑制自身多聚体化而使其处于非活化状态,在NOD结构域内由HD1、WHD、HD2形成一个特殊的ADP结合囊,稳定NLRC4的非活性构象。当有细菌感染等活化信号出现时,LRR结构域识别配体,引起自身构象变化,促进ADP转化为ATP,解除LRR所介导的对NOD结构域寡聚化的抑制,触发寡聚体化继而暴露出CARD效应结构域,NLRC4炎性小体被激活,引起caspase-1的活化,触发炎症反应[6]。
革兰阴性致病菌,如鼠伤寒沙门菌、弗氏志贺菌、铜绿假单胞菌、嗜肺军团菌等,可通过Ⅲ型分泌系统(type three secretion system,T3SS)的跨膜膜针状结构将其鞭毛蛋白或PrgJ样杆状蛋白导入宿主细胞内,激活NLRC4炎性小体,导致巨噬细胞死亡和促炎细胞因子释放[7]。与NLRP3和AIM2不同,NLRC4不直接与细菌配体相互作用。NLR家族凋亡抑制蛋白(NLR family apoptosis inhibitory protein,NAIP)家族是具有3个杆状病毒抑制凋亡蛋白重复域(baculovirus inhibit apoptosis protein repeat domain,BIR)结构域的NLR,其作为配体受体介导NLRC4炎性小体活化。相关的体外实验和小鼠体内实验均表明,NAIP1可与T3SS的针状蛋白PrgI结合,NAIP2可特异性地结合T3SS的基座蛋白PrgJ,NAIP5和NAIP6可结合细菌的鞭毛蛋白,并进一步招募NLRC4组装形成NAIP/NLRC4炎性小体复合物,激活caspase-1介导的炎症反应[8-9]。
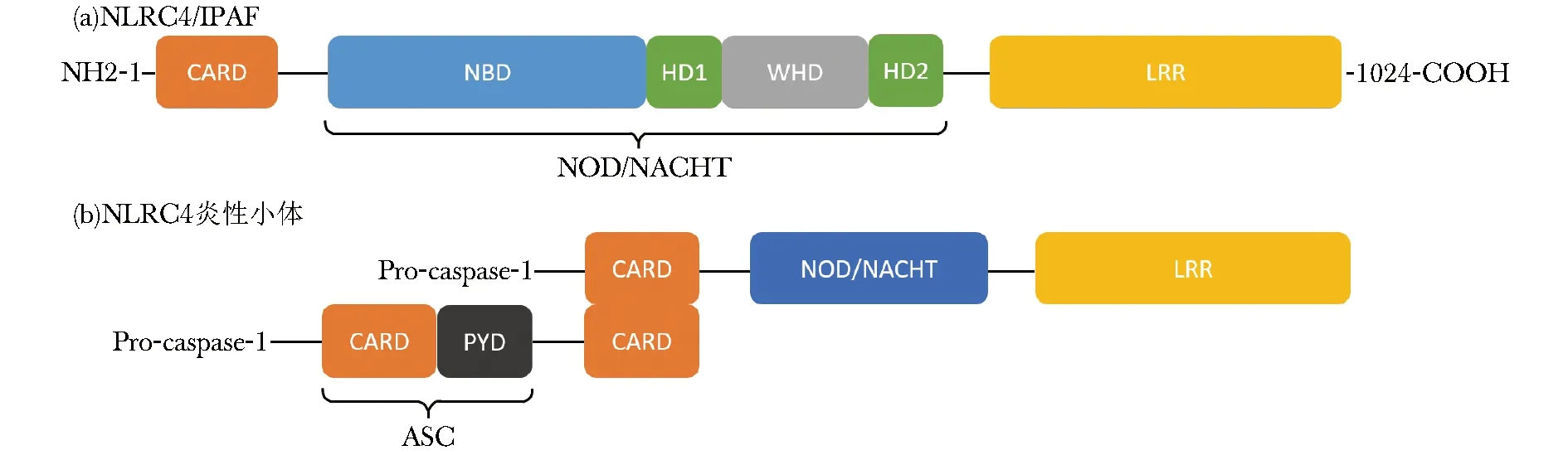
图 1 LRC4和NLRC4炎性小体结构示意图
Fig 1 NLRC4 and NLRC4 Inflammasome
IPAF:IL-1β转换酶蛋白激活因子;CARD:caspase募集结构;NBD:核苷酸结合域;WHD:翼状螺旋结构域;HD:螺旋结构域;LRR:亮氨酸重复结构域;PYD:热蛋白结构域
NLRC4炎性小体活化后产生两种效应(图2)。一方面,NLRC4炎性小体可通过ASC或NLRC4蛋白的CARD结构域募集pro-caspase-1,在CARD-CARD相互作用下使两个相邻的pro-caspase-1发生自身水解,产生具有活性的caspase-1,进一步剪切pro-IL-1β和pro-IL-18,分泌成熟的IL-1β和IL-18,引起炎症反应。另一方面,caspase-1可诱导细胞焦亡(pyroptosis)。细胞焦亡是一种细胞程序性死亡,表现为细胞胀大,最终导致胞膜破裂、细胞内容物释放,引起强烈的炎症反应。在所有炎性caspase中(包括caspase-1)存在共同的底物蛋白消皮素D(Gasdermin D,GSDMD),它属于一个名为gasdermin的蛋白家族,该家族还包括GSDMA,GSDMB,GSDMC,DFNA5(GSDME),DFNB59等。炎性caspase通过切割GSDMD释放其N端结构域,该结构域可以识别并结合细胞膜上的磷脂类分子,在胞膜上形成孔道,导致细胞渗透压变化,最终引发细胞焦亡[10]。细胞焦亡除了能引起宿主的炎症反应,还能通过胞膜孔道捕获致病菌,防止细菌感染其他细胞,促进宿主启动免疫防御清除致病菌[11-12]。
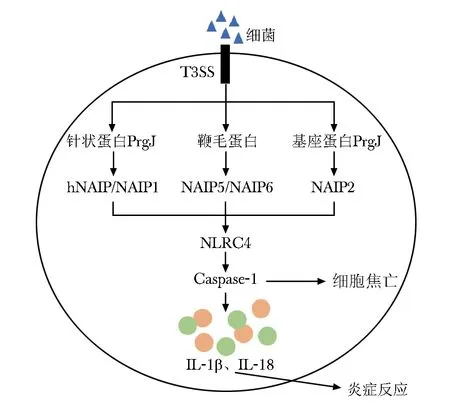
图 2 NLRC4炎性小体的活化机制Fig 2 Activation mechanism of NLRC4 inflammasomeT3SS:Ⅲ型分泌系统;NAIP:NLR家族凋亡抑制蛋白;hNAIP:人NLR家族凋亡抑制蛋白
3 NLRC4炎性小体相关疾病
3.1 NLRC4炎性小体病——3种表型:AIFEC、NOMID、FCAS4
自身炎症性疾病是一种系统性或器官特异性炎症,大多是由固有免疫通路失调和过度活跃引起的,并且多为单基因疾病[13]。目前研究较多的主要有3种表型,分别为自身炎症伴婴儿小肠结肠炎(autoinflammation with infantile enterocolitis,AIFEC)、新生儿多系统炎症性疾病(neonatal onset multi-system inflammatory disease,NOMID)和家族性寒冷性自身炎症综合征4(familial cold autoinflammatory syndrome 4,FCAS4)[14]。AIFEC和NOMID与NLRC4基因HD-1和NBD亚结构域变异相关(S171F,T177A,T337S,T337N和V341A),而FCAS4与WHD亚结构域变异(H443P和S445P)相关(图3)。据报道,WHD结构域的变异可能会不同程度地促进caspase-8介导的细胞死亡,而HD-1的变异则不会[15]。NLRC4突变促进了NLRC4炎性小体的异常激活、IL-1β和IL-18的产生以及巨噬细胞的死亡,其具体机制有待进一步研究。
2014年的两项研究最先报道了一组NLRC4炎性小体病,阐述了NLRC4功能性获得性突变是婴儿小肠结肠炎和反复发生巨噬细胞活化综合征(macrophage activation syndrome, MAS)的病因,并与新生儿死亡有关[16-17]。后来人类孟德尔遗传数据库(Online Mendelian Inheritance in Man,OMIM)将这类疾病命名为AIFEC。AIFEC是一种慢性炎症性疾病,伴有可危及生命的超急性发作,主要表现为发热、分泌性腹泻,以及外周血细胞计数降低、肝功能障碍、凝血功能障碍、血清铁蛋白显著升高和骨髓噬血细胞现象等MAS表现,并有血清IL-18明显升高(常>104 pg/ml)[14, 17-18]。
NOMID属于冷炎素相关周期性综合征(cryopyrin-associated periodic syndromes,CAPS)中的一组疾病,既往认为与NLRP3基因突变有关。NOMID的主要临床表现为发热、皮疹(尤其是荨麻疹)、关节炎、中枢神经系统炎症(感觉神经性聋、无菌性脑膜炎、脑积水、脑萎缩、智力发育迟滞等)。2017年,Kawasaki等[19]报道了第一例体细胞NLRC4突变所致与NOMID临床表现一致的病例。目前尚未有其他病例报道。
FCAS1是另一组属于CAPS的疾病,同样与NLRP3基因突变有关,但临床表现较NOMID更加轻微,主要表现为遇冷发生的一过性发热、荨麻疹、关节痛等症状。2014年,Kitamura等[20]曾报道过一个存在NLRC4基因突变的家系,突变患者的临床表现为新生儿发热、感冒诱发的荨麻疹和关节炎,患者的细胞在体外寒冷环境下分泌IL-1β明显增多,IL-18的水平未检测。由于该组NLRC4基因突变患者临床表现与NLRP3相关的FCAS1有相似的临床表现,因此被称为FCAS4。2017年,另一项研究报道了以皮肤红斑结节、荨麻疹、关节痛和晚发性肠胃炎为主要症状的一组病人,在这组病人中发现了一种新的NLRC4基因突变S445P,并且该组病人的IL-18明显升高[21]。该研究也首次对NLRC4功能性获得性突变患者皮肤病变的组织病理学进行了描述,皮肤活检显示真皮淋巴细胞-组织细胞浸润。
在缺乏针对 NLRC4基因突变的特异性治疗的情况下,目前主要以下游炎症介质作为治疗靶点。经研究已证实有效的药物有重组IL-1受体拮抗剂阿那白滞素(anakinra)、糖皮质激素、环孢素A、静脉输注丙种球蛋白,现正在进行临床试验的药物有重组IL-18结合蛋白和抗IFN-γ单克隆抗体[19,21-22]。
3.2 细菌感染
NLRC4炎性小体在促进肠道细菌病原体的清除方面起着重要作用。Sellin 等[23]的研究显示,在小鼠肠上皮细胞中,缺失NAIP1-6导致感染早期沙门氏菌的细菌负荷增加,说明感染的肠上皮细胞排出沙门氏菌依赖于NAIP蛋白在肠道中的表达。对特异性敲除NLRC4的肠上皮细胞的研究证明,NLRC4通过激活caspase-1和caspase-8介导细胞程序性死亡,促进感染性肠上皮细胞的排出[24]。然而,Chen等[25]发现,与巨噬细胞和肠上皮细胞不同,沙门氏菌感染小鼠中性粒细胞后并不发生细胞焦亡,而是在感染部位持续产生IL-1β,使促炎作用和抗菌效应最大化。另外,Hermansson等[26]研究发现,志贺菌感染上皮细胞、巨噬细胞、中性粒细胞、树突状细胞、T/B淋巴细胞等不同的细胞后,均可调控炎性小体活化的“开关”,抑制炎性小体的活化,发生免疫逃避,导致细菌感染。
嗜肺军团菌可经呼吸道吸入引起军团菌肺炎。嗜肺军团菌的鞭毛蛋白为NLRC4炎性小体的激活因子,在NAIP5和NLRC4协同作用下诱导caspase-1的活化[27]。同时也有研究发现,在低嗜肺军团菌载量下,caspase-1的活化依赖于鞭毛蛋白和NLRC4;而在高细菌载量下,上皮细胞中caspase-1的活化不依赖NLRC4,表明NLRC4炎性小体在caspase-1活化中的作用可能与细菌载量有关[28]。最近的研究表明,肿瘤坏死因子(tumor necrosis factor, TNF)和活性氧(reactive oxygen species, ROS)也在控制嗜肺军团菌感染的固有免疫过程中发挥重要作用,而并不依赖于NLRC4和caspase-1[29]。
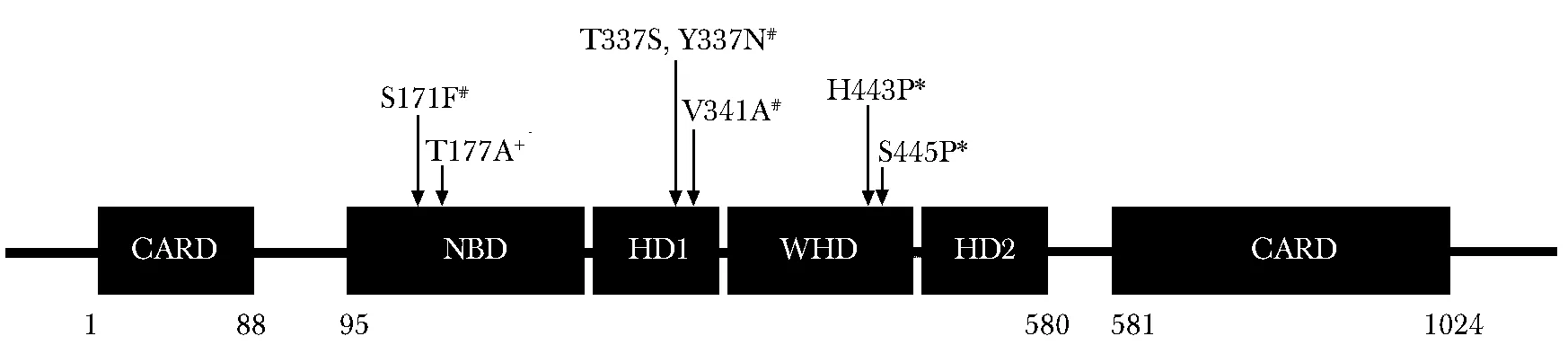
图 3 与NLRC4炎性小体病相关的NLRC4基因突变位点示意图
Fig 3 NLRC4 gene mutation sites associated with NLRC4 inflammasomopathies
#:自身炎症伴婴儿小肠结肠炎相关的突变;+:新生儿多系统炎症性疾病相关的突变;*:家族性寒冷性自身炎症综合征4相关的突变
铜绿假单胞菌是一种医院内常见的条件致病菌。Lindestam等[30]发现,铜绿假单胞菌的菌毛蛋白也可能是NLRC4炎性小体的一种激活因子。据报道,在低细菌载量时,鞭毛蛋白激活NLRC4参与铜绿假单胞菌感染后的caspase-1活化;但在高细菌载量时,缺乏鞭毛蛋白的铜绿假单胞菌突变体仍能够通过NLRC4活化caspase-1,其机制暂不明确。
3.3 其他疾病
近年来的研究揭示炎性小体激活可以导致神经元和胶质细胞死亡,造成缺血性卒中后脑损伤和神经功能缺损。Poh等[31]的研究证实了小胶质细胞在体外和体内缺血条件下可释放NLRC4炎性小体成分和促炎细胞因子(如IL-1β、IL-18),介导炎症反应、细胞凋亡和细胞焦亡,使用抑制caspase-1的药物能够有效抑制细胞死亡,表明NLRC4炎性小体在小胶质细胞和脑组织中表达、组装或激活的途径可能是卒中的潜在治疗靶点。
炎性小体成分的基因突变可能增加人类对癌症的易感性。一项研究表明NLRC4通过抑制细胞增殖和促进细胞死亡来防止结直肠癌的发生[32],但未在另一项研究中得到证实[33]。NLRC4还可以放大巨噬细胞内的炎症信号通路,并促进CD4+和CD8+T细胞分泌IFN-γ,抑制黑色素瘤小鼠的肿瘤生长[34]。另有研究发现,NLRC4炎性小体激活引起的IL-1β分泌,可促进脂肪细胞介导的血管内皮生长因子A表达,促进血管生成,加速乳腺癌的进展[35]。
NLRC4缺乏可减缓糖尿病肾病进展,表现为血糖降低、尿白蛋白排泄减少。一项研究显示,野生型糖尿病模型小鼠肾脏改变为肾小球肥大、肾小球基底膜增厚、细胞外基质积累,所有这些效应在NLRC4缺陷小鼠中都得到了显著的缓解,证明NLRC4炎性小体活化水平的降低减少了肾脏的炎症反应和巨噬细胞的累积,并下调NF-κB和JNK信号通路的表达,改善了肾脏损伤[36]。
4 总结
NLRC4炎性小体不仅在防御细菌感染的固有免疫应答中发挥作用,也与自身炎症性疾病密切相关。但仍有许多问题待进一步研究,如:NLRC4炎性小体病的活化因子是什么,NLRC4基因突变引起的NLRC4炎性小体异常活化的机制是什么,是否还有其他的活化因子和活化途径,NLRC4炎性小体在不同病灶部位的具体作用机制,NLRC4炎性小体激活后是否产生caspase-1介导的炎症反应和细胞焦亡以外的效应,NLRC4炎性小体与其他炎性小体的相互作用等。对NLRC4炎性小体的作用机制的研究将为相关疾病的发病机制提供证据,并为疾病的预防和治疗提供新方向。
