大豆调和油氧化酸败后的组分变化
2019-06-25刘志明类彦波孙清瑞赵建波李秋兵樊云雪黄蕊蕊
刘志明,类彦波,孙清瑞,赵建波,李秋兵,樊云雪,黄蕊蕊
(1.黑龙江八一农垦大学食品学院,黑龙江大庆 163319;2.大庆市食品药品检验检测中心,黑龙江大庆 163319)
大豆油脂肪酸(Fatty acid,FA) 由饱和脂肪酸(Saturated fatty acid,SFA) 与 不 饱 和 脂 肪 酸(Unsaturated fatty acid,USFA) 构成,且USFA含量很高[1-3]。USFA具有调节生理机能、增强机体健康、预防多种疾病等功能,尤其能预防心血管疾病、神经或精神类疾病、内分泌失调及代谢综合症类疾病,还能抗炎和抗癌等。在减缓记忆力衰退、防止皮肤老化、延缓衰老、抗过敏反应及促进毛发生长等方面也有积极作用[4-5]。大豆油是我国主要的食用植物油,既以纯油食用,又常与其他食用植物油复配成大豆调和油(Blended soybean oil,BSO),以改善营养结构与风味。BSO中亚油酸和亚麻酸等USFA含量因原料油不同而有较大差异,但其氧化稳定性较差的属性未变。大豆油的USFA在其贮存过程中,受外界因素(氧气、温度、光照、水分、酶、金属离子等)影响易发生氧化酸败[6-10],产生的初、次级产物劣化风味,影响健康,乃至引发疾病[11-13]。其氧化初期形成不稳定氢过氧化物,继而分解成次级产物,如醇、醛、酮、酸、环氧化物或生成聚合物等[14-15],产生的过氧化物与自由基,进入人体可引起衰老、癌症及其他有关疾病[16-17],加大其贮藏、运输、消费等过程中的保质难度。因此,对部分模拟贮存条件的加速氧化酸败BSO试样进行综合仪器分析,弄清BSO的FA和USFA含量变化,以及由氧化酸败产生的有害物质情况,可对BSO贮存期内的食品安全状况提供有益信息,也为全面分析与正确判断BSO等食用植物油的氧化酸败情况提供参考。
1 材料与方法
1.1 材料与仪器
大豆调和油:SFA MUFA PUFA 为 (0.2~0.3)(0.6~1.0) 1.0;37组分脂肪酸甲酯混合标准品(C4~C24);三氟化硼(14%甲醇溶液)、甲醇,色谱纯;氢氧化钠(AR)、正己烷(GC),天津市光复精细化工研究所提供;氯化钠(AR),杭州嘉辰化工有限公司提供;无水硫酸钠(AR),天津市大茂化学试剂厂提供。
7890A GC system,安捷伦科技有限公司产品;TRACE 1300 ISQ LT单四极杆气相色谱质谱仪联用仪(Gas Chromatograph-ISQ LT Single Quadrupole Mass Spectrometer),赛默飞世尔科技公司产品;傅里叶变换红外光谱仪(FTIR),珀金埃默仪器有限公司产品。
1.2 试验方法
1.2.1 试样氧化
模拟室内照度(840 lx)和相对湿度(40%),控制氧化温度50℃,氧化试样容器初始顶空率50%,氧分压(Oxygen partial pressure,OPP) 分别设为5.3,10.5,21.0,42.0,84.0 kPa,进行BSO氧化试验,累计氧化79~87 d,直至感评其彻底腐败,期间多次测定试样各种性能参数(等量取样,保持顶空率不变),探究其氧化规律(另著文)。选取经84.0 kPa累计氧化1 918 h的氧化试样,将其与原始试样一并甲酯化,同时进行GC,GC-MS和FTIR分析,对比BSO氧化前后试样的特征组分,确定有害物质。
1.2.2GC与GC-MS分析
(1) 试样甲酯化。参照文献[18-19]方法,甲酯化BSO原始样及其氧化试样。用滴管向50 mL圆底烧瓶中加2滴试样、10 mL浓度0.5 mol/L的氢氧化钠甲醇溶液,于85℃条件下回流10 min,用移液枪加入三氟化硼甲醇溶液7 mL,继续回流5 min。取出后冷却,加入2 mL正己烷(萃取),摇动,加入适量饱和氯化钠溶液,轻摇,促进体系分层。继续加入氯化钠饱和溶液至烧瓶颈部,静置,吸出上层溶液,放入5 mL具盖塑料样品杯中,加入3 g无水硫酸钠粉末,盖上盖子,手动振摇1 min,静置5 min(脱水),用有机针式滤膜过滤,取适量放入样品瓶,密闭,冷藏备用。
(2) GC分析。参照GB 5009.168—2016[20]中5.3法对比分析甲酯化试样和混合脂肪酸标样。色谱柱:Agilent DB-FFAP毛细管柱,载气:氮气(99.99%),1.0 mL/min,进样口温度260℃,分流比10∶1,检测器温度280℃,柱温初温140℃,保持5 min,再以4℃/min升温至240℃,保持15 min,进样量0.5 μL。
(3) GC-MS分析[19-20]。①GC分析。对比混合脂肪酸标样确定甲酯化试样的FA组分。Agilent DB-5型毛细管柱:柱长30 m,内径0.25 mm,膜厚0.10 μm,固定相为5%苯基-甲基聚硅氧烷;进样口温度280℃;柱温初温150℃,保持5 min,再以3℃/min升温至170℃,保持5 min,再以3℃/min升温至210℃,保持2 min,最后再以10℃/min升温至260℃,保持5 min;试样进样量为1 μL,分流比100∶1,载气为氦气(99.99%),流速1.2 mL/min。②MS分析。对甲酯化试样进行全离子扫描,得GC-MS总离子流图,同时分析混合脂肪酸标样,利用标样谱图对比解析试样谱图及其各保留时间对应的FA组分,并用峰面积归一化法求算FA相对含量(Rrelative content,RC)。传输线温度250℃,EI离子化模式,离子源温度230℃,电子能量70 eV,全扫描模式,分子离子扫描范围50~650 m/z,溶剂延迟5 min。
1.2.3 FTIR分析
向一片干净的KBr晶片中心滴1滴试样,在其上叠放另一干净的KBr晶片,小心转动晶片,使试样形成均匀薄膜。将叠放的KBr晶片装入可拆式液体池中,固定好,放在红外光谱仪样品支架上,在设定条件下分析试样,得其红外吸收光谱图,分析确定BSO中所含物质。
2 结果与分析
2.1 GC分析结果
BSO试样的GC谱图见图1,BSO氧化试样的GC谱图见图2。
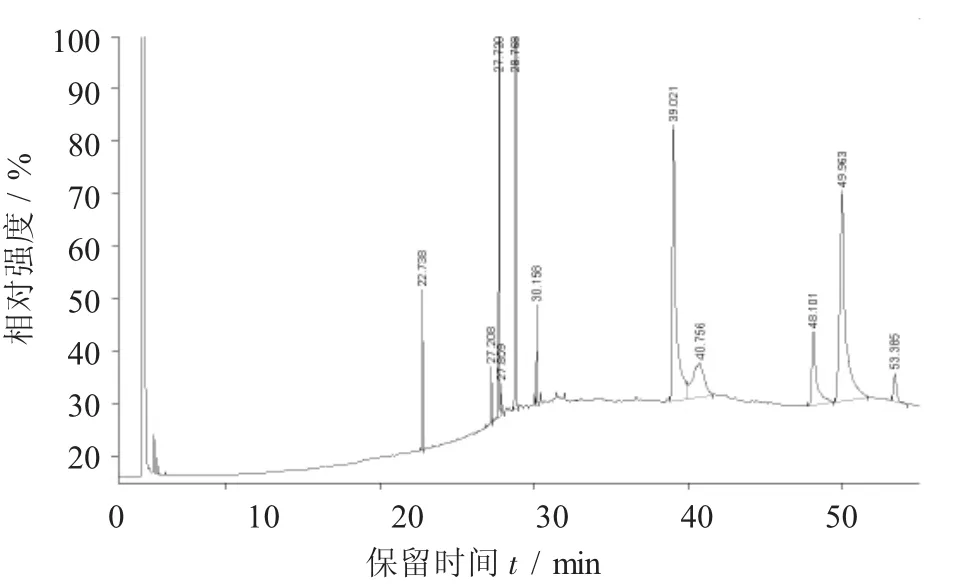
图1 BSO试样的GC谱图
对比混合脂肪酸标样谱图,并参照GB 5009.168—2016中图B.1和表C.1,得出表1分析结果。表1数据表明:①BSO试样经长时间氧化后,SFA的 RC由 26.09%增至 37.56%,C16∶0,C17∶0,C18∶0的RC分别增加了1.34%,4.45%,5.68%。②USFA的RC由73.91%降至62.07%,且主要USFA的RC显著降低。C18∶1n9t+C18∶1n9c的RC由25.28%降至20.86%,并且C18∶2n6t+C18∶2n6c的RC由17.03%降至6.01%。其中,C18∶2n6t的RC由9.20%至消失,C18∶3n6的RC由27.85%降至25.35%。③其他USFA的RC略增,C16∶1,C17∶1,C18∶3n3的RC分别由1.10%,0.68%,1.98%变至1.59%,0.79%,7.46%,前两者增幅不大,而后者增幅很大,是否发生了结构改变或与氧化后C18∶2n6t的消失有关,尚不清楚,也可能是GC法局限性所致。这些情况揭示BSO在试验条件下经1 918 h氧化后,FA组分发生了显著变化,且饱和化严重,仅靠GC法确定FA构成尚有不确定性。

图2 BSO氧化试样的GC谱图
2.2 GC-MS分析结果
BSO试样及其氧化试样的GC分析结果见表1,BSO试样GC-MS分析结果见表2。
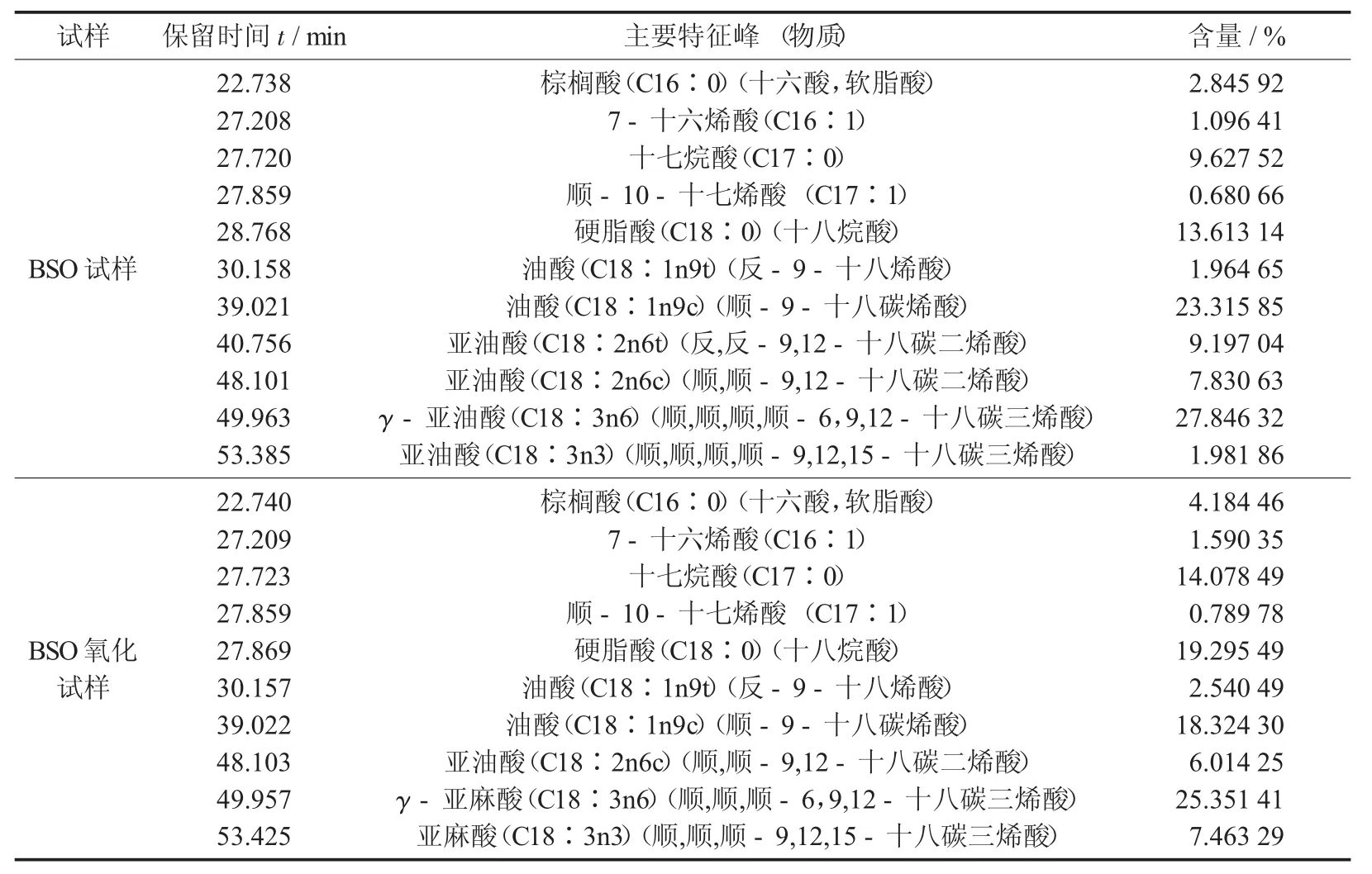
表1 BSO试样及其氧化试样的GC分析结果

表2 BSO试样GC-MS分析结果
数据表明:①BSO试样氧化后,FA由6种增至10种,较原来的多了C18∶2n6t,C22∶0,C20∶0,C20∶1n11c。氧化酸败过程使BSO的FA组成与结构发生了改变,且令FA高级化。②SFA中C16∶0和C18∶0的RC无明显变化,因其属于稳定FA。③USFA中亚油酸的RC略减,且产生反式亚油酸(一般认为高温是食用植物油加工、含油食品加工时USFA反式异构化主要原因[21],未见试验温度下发生异构化的报道),若果真如此,机理不明,且会增加食品安全风险[22]。油酸的RC略有增多,亚麻酸含量基本无变化。这些FA在BSO中是主要成分也是发生氧化酸败的主体,且变化较为复杂。④不同的分析方法得到的FA的RC差异较大。C18∶1n9t在表1中由1.96%升至2.54%,明显增多,而其在表2中几乎无变化;C18∶2n6t在表1中由1.92%而消失,在表2中却由无到0.30%;C18∶3n3在表1中由1.98%增至7.46%,在表2中几乎无变化。FA的这些差异仍难解释。但就分析方法而言,GC-MS分析得到的表2数据应比GC分析得到的表1数据更可靠。⑤不能孤立地分析BSO的氧化酸败过程,FA变化还需用其他方法协同分析加以解释。理论上,氧化酸败过程能使USFA饱和化,还可使初级氧化产物(过氧化物)分解为较小分子的次级产物,如酸、醇、醛、酮等。因GC-MS法仍是以匹配度为依据的分析方法,对于结构相近的同系物、异构体等,因质谱图相似,可能造成检索结果的不可靠,故难以全面、准确地判定试样氧化酸败产物及其含量,上述仅从FA组分与含量角度的分析只能部分地反映BSO氧化酸败状况,并不能反映其氧化酸败的全貌,若要从结构变化入手描述BSO试样的氧化酸败,还应借助于FTIR分析。
2.3 FTIR分析结果
BSO试样的FTIR谱图见图3,BSO氧化试样的FTIR谱图见图4。
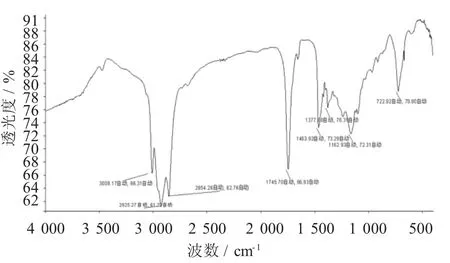
图3 BSO试样的FTIR谱图
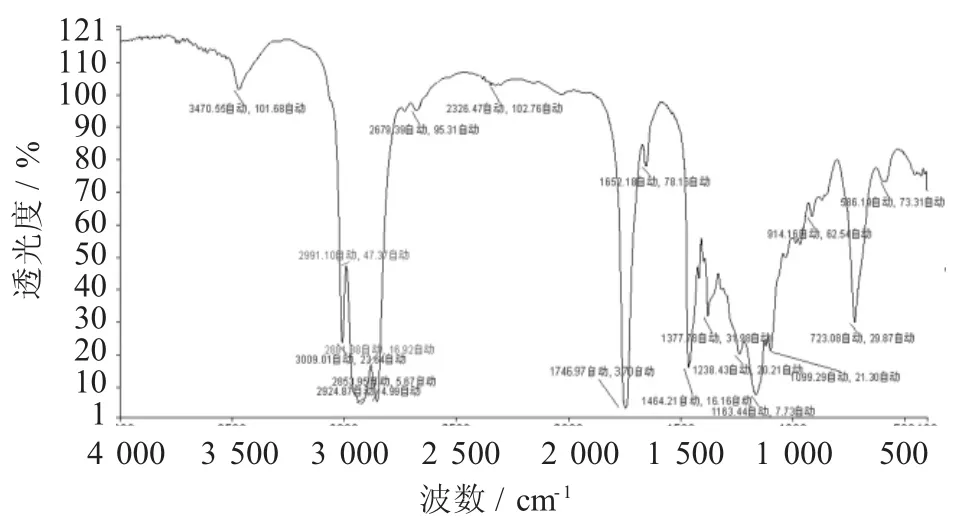
图4 BSO氧化试样的FTIR谱图
综合分析表明:①BSO原始试样的FTIR谱图只有8个峰,其试样氧化后谱图增至18个峰,官能团数目大大增加,说明氧化产生了多种化合物;②BSO原始试样谱峰以烯烃中次甲基(3 009.17 cm-1),烷烃中甲基、亚甲基(2 925.27,2 854.28,1 463.93,1 377.80 cm-1)的C-H振动为主。在1 745.70 cm-1处有酯基或羧基中的C=O伸缩振动;1 162.93 cm-1处有甘油三酯中C-O,(RCO)2O中C-O,-COOR中C-O-C的伸缩振动,R-O-R'中C-O的不对称伸缩振动;722.92 cm-1处碳链的骨架振动、长碳链烷基-CH2-的面内摇摆振动、长链-(CH2)n-(n≥4)的面外弯曲振动、-(CH2)n-链中-CH2-的C-H变形振动、顺式-CH=CH-中C-H的面外弯曲振动、顺式-CH=CH-中C-H键的弯曲振动,而其氧化试样中除了有甲基、亚甲基(2 991.10,2 925.27,1 377.78 cm-1)、烯基(3 009.17,1 652.18,7 22.92 cm-1)、叔丁基(1 464.21 cm-1)的官能团振动峰外,还有酮(1 652.18 cm-1)、醛(914.16 cm-1)、酯(3 470.55,1 745.70,1652.18cm-1)和羰基(2 326.47 cm-1),羧基(1 652.18,1 238.43,914.16 cm-1)及羧羰基(1 745.70,1 652.18 cm-1),醚基(1 162.93 cm-1),酸酐基团(1 163.44,1 099.29 cm-1),醇基(1 238.43,1 099.29 cm-1)等官能团的振动峰,这些官能团极性较强,其相应化合物可使氧化试样κ增大,同时也是BSO试样氧化的重要标志。其中许多化合物也是引发潜在食品安全问题的有害物质,抑制或避免食用油品质下降及此等有害物质的产生也是油脂贮藏应解决的难题。我国食用植物油销量大、种类多[23],其保质贮存是关乎食品安全的大事。
2.4 相关讨论
食用植物油的脂肪酸和活性物质的种类与含量都影响其氧化稳定性[24],试验用BSO产品的配料油的生产工艺、精炼程度,BSO是否加入抗氧化剂(包装标签标注的配料中有“食品添加剂”字样,可能为抗氧化剂)等都对试验结果(与BSO贮藏稳定性相关) 有影响[9,25-26]。试样氧化温度与OPP远高于一般贮存及消费情况,相对湿度和温度均为常见情况,故可认为高温和高OPP是引起试样氧化酸败的主因。BSO贮存时,温度可控度较大,OPP理论上可通过气调或真空包装等方式加以控制[27],但仅限于贮存期,消费期宜采用特定隔氧容器包装。试样氧化条件及氧化状态(氧化条件控制状态与程度、试样的均匀程度、氧化酸败结果的再现性等)可能影响分析结果,试样前处理方法的差异也可能对结果产生影响[4],这些方面还有待深入研究。
3 结论
BSO试样氧化酸败的结果使SFA增多,USFA减少,FA组分变化较大,严重损害其营养功能和风味,试验条件下已严重哈变。BSO试样中FA的变化与很多因素(BSO调配比例、氧化试验条件与方法、FA等组分的分析条件与方法等)相关,其FA的RC既由各自绝对量变化决定,也受各FA的RC“此消彼长”的影响,只是特定试验条件下的结果。BSO试样在高OPP和较高温度加速模拟试验条件下的深度氧化酸败,产生了酮、醛、酯等羰基化合物,游离羧酸、酸酐,醚、醇等物质,其品质严重下降,继续食用将引发食品安全问题。加速模拟试验条件虽然与通常食用植物油的生产、贮存、运输、销售、消费的情况不完全一致,发生氧化酸败的速率、程度和产物也不尽相同,但发生的相关反应及产物的种类基本相同,反映出食用植物油贮藏期间氧化酸败的事实。以GC,GC-MS和FTIR耦合分析技术能全面、准确地揭示破坏性氧化酸败试验中BSO试样的FA变化及次级产物的组成与结构情况,而单独以GC或GC-MS中的1种或2种都难以达到满意的表达效果,甚至得出错误结论。
