酸催化丙酮碘化反应机理的理论研究
2019-06-25陈喜陈慧慧陆娟凤
陈喜, 陈慧慧,陆娟凤
(中南民族大学 化学与材料学院,武汉430074)
羰基化合物α-位的卤代反应是一类重要的有机反应[1, 2].通过在羰基化合物的α-位引入卤素原子,可使分子的生化性质发生变化,成为具有某种特定生物活性的药物分子.同时,α-位的卤化还可激活羰基化合物的反应活性,使其更容易发生官能团的交换.因此,该类化合物被广泛应用于医药或农药中间体的制备与合成.由于卤素的浓度便于检测,且反应速度适中,此类反应也常被用于教学研究.
大量实验表明,羰基化合物α-位的卤代反应可在多种化学条件下进行[2,3].根据反应条件的不同,这些反应大致可分为酸催化、碱催化、金属催化和自由基反应等类型.其中,对酸催化条件下丙酮的碘化反应研究较为深入.根据实验数据,提出了该反应的一般机理(图1)[2, 4, 5].由图1可知:酸催化丙酮碘化反应的第一步是丙酮的羰基从H3O+上获得质子,形成高能中间体(CH3)2COH+.经过分子内电子转移,该中间体的羰基碳上带上了1个单位的正电荷,这将活化相邻的2个甲基,使其上的氢原子具有部分酸性.第二步中,溶液环境中的1个水分子从中间体(CH3)2COH+的1个甲基上拔去1个质子,生成另一种高活性中间体——丙烯醇.最后,该丙烯醇与碘分子进行加成-取代反应,生成目标产物碘化丙酮.

图1 酸催化丙酮碘化反应机理Fig.1 Reaction mechanism of acid-catalyzed iodation acetone
根据实验数据,该反应的速率方程为:
v=k[A] [H+],
(1)
式(1)中的v,k,[A]分别代表反应速率、速率常数及反应物丙酮的浓度.由式(1)可见:反应速率取决于反应物丙酮和酸催化剂的浓度,而与反应物碘的浓度无关,这意味着速度控制步中只有丙酮和酸参与反应,而碘没有参与.结合图1中的机理,可见反应(1),即丙酮的质子化步骤,为酸催化丙酮碘化反应的决速步骤.以上机理被广泛接受,但仍存在矛盾之处,试分析如下:
(1)图1给出的三步机理中,第一步为羰基氧获得质子,第二步为甲基碳失去质子.决速步若为第一步,意味着氧得失质子较碳得失质子更困难,与一般认识相悖.
(2)反应在水溶液中进行,前两步反应中的质子得失转移发生在相邻的两个基团与水分子之间.因此,环境水形成的氢键网络可能使这两种质子转移同时发生,这种协同的质子转移将有助于分散体系所带的正电荷,降低反应的能垒.
以上分析表明:图1中提出的机理可能不是酸催化条件下丙酮碘化反应的最佳途径.由于该反应具有代表性,有必要对其机理进行重新研究,获得该反应的基本途径和热力学性质,用于指导研究其他酮类的α-位卤代反应. 随着计算技术的发展和硬件水平的提高,高精度的量子化学方法已被广泛应用于分子的电子结构[6]和反应机理等研究领域. 为此,本文综合运用多种量子力学方法研究了该反应的多条竞争性途径,计算了各途径的热力学能参数,从微观的角度阐明酸催化条件下丙酮碘化反应的基本途径,以深入理解和认知此类反应.
1 计算方法
研究工作所涉及到的反应物、过渡态、中间体和产物结构均用GaussView软件进行构建.对于所构建的初始结构,采用量子化学密度泛函理论[7]B3LYP方法[8-10]和混合基组进行几何构型优化.混合基组中,对碘元素适用MiDiX基组,对其他类型元素则适用6-31++G(d, p)基组.随后,在相同水平上对所有的优化结构进行振动频率分析计算,确认这些结构是反应势能面上的能量极小值点或鞍点,同时估算这些分子结构298 K下内能和吉布斯自由能的热力学能校正,记为ΔΔE(therm, 298 K)和ΔΔG(therm, 298 K).对于所有过渡态结构,还进行了内禀反应坐标(IRC)计算以确认过渡态与各极小值点的对应.鉴于B3LYP方法对色散作用考虑不充分,本文采用M062X方法[11]和6-311++G(2d,2p)基组(对碘元素适用MiDiX基组)对前面得到的优化结构进行了单点能计算,以获得较为准确的电子结构能量Eele.
由于反应时在水溶液中进行的,须充分考虑溶剂环境对该反应的影响.为此,选取SMD溶剂化模型[12, 13],使用B3LYP方法和6-31+G(d)基组(对碘元素适用MIDIX基组)计算了前面工作中得到的分子结构在298 K下水溶液中的能量,以此为基础计算这些结构的溶剂化能ΔΔG(sol, 298 K).水溶液中的反应物、过渡态、中间体和产物的自由能G(sol, 298 K)按照公式2进行估算.
G(sol, 298 K)=Eele+ΔΔG(therm, 298 K)+ΔΔG(sol, 298 K) .
(2)
所有结构优化、单点能以及溶剂化计算均使用Gaussian09程序,在64位Linux系统机群上完成.
2 结果与讨论
研究表明:丙烯醇是酸催化丙酮碘化反应的中间体[2],据此可将反应大致分为丙酮烯醇化和烯醇碘化2个反应阶段,计算研究也是针对这2个反应阶段展开的.在2个反应阶段,均存在多条竞争反应途径,现分述如下.
水溶液中丙酮的烯醇化存在5条竞争性反应途径(见图2),虽然前3条途径的反应物中无水合质子参与,但也有助于理解水合质子在烯醇化阶段的作用.

a)直接质子转移;b)单水辅助质子转移;c)二水辅助质子转移;d)酸辅助分步质子转移;e)酸辅助单步质子转移图2 丙酮转化为烯丙醇的5条竞争反应途径Fig.2 Five competitive pathways for conversion from acetone to allyl alcohol
首先研究了丙酮的α-氢直接转移到羰基氧上的反应途径(见图3a).由图可见:常温条件下此步反应的自由能能垒高达66.87 kcal/mol,意味着丙酮的α-氢直接转移到羰基氧上困难.这种困难主要是过渡态TS1中过大的四元环张力造成的.
在丙酮的羰基和甲基之间引入1个水分子可以减小过渡态结构中的环张力(见图3b).由图可知:该水分子直接参与了反应,通过“接力”的方式将质子由甲基转移到羰基氧原子上;在反应的过渡态结构TS2中,参与“接力”的基团与水分子形成一个闭合的六元环,其环张力远小于前述四元环张力.相比于直接质子转移,这种单水辅助质子转移途径的自由能能垒减少了22.59 kcal/mol.若引入额外的水分子到反应体系(见图3c),则能进一步稳定过渡态结构的能量.在过渡态结构TS3中,2个水分子、羰基与甲基之间形成了一个松弛的八元闭合环,几乎消除了张力的影响,相应的自由能能垒降至33.72 kcal/mol (25 ℃),但该数值仍远高于室温下能顺利发生的化学反应的活化能阈值(~20 kcal/mol).因此,室温下仅仅依靠水分子完成酮-烯醇的转化依然困难.
众所周知,增加溶液的酸性有助于酮向烯醇的转化.但酸催化烯醇化反应的机理(见图1)存在矛盾,必须重新进行检验.

a)直接质子转移途径;b)单水辅助质子转移途径;c) 二水辅助质子转移途径图3 丙酮烯醇化反应的直接质子转移及水辅助质子转移途径的分子结构、相对自由能Grel及部分几何参数Fig.3 Molecular structures, relative free energiesGrel and partial geometric parameters for direct proton transfer and water-assisted proton transfer pathways of enolization of acetone
探索性计算表明,H3O+必须在水分子的协助下才能发挥其催化作用.通过计算发现了2条不同的水辅助H3O+催化丙酮烯醇化反应途径(见图4a和4b).图4a中的途径是一个两步反应步骤构成(羰基质子化和碳上拔氢),与传统机理一致.能量计算表明,碳上拔氢步骤过渡态TS5的自由能大小为23.95 kcal/mol,而羰基质子化步骤过渡态TS4的自由能仅为7.72 kcal/mol,充分说明从甲基上拔去质子的难度要远远大于羰基氧接收1个质子的难度.反应中水分子形成了较强的闭合的氢键网络,在分散体系正电荷、稳定反应过渡态方面起到了重要作用.
进一步反应坐标计算表明,羰基质子化和甲基失氢可通过一个反应完成(见图4b). 由于转化过程水分子形成的氢键网络平衡了电荷的分配,同时缓解了张力带来的影响.与直接α-氢转移相比,此途径的自由能能垒减少了约50 kcal/mol,降至16.97 kcal/mol.
比较以上5条反应途径,发现第5条途径的自由能能垒值(16.97 kcal/mol)最低,较另4条途径的自由能能垒值低约7~50 kcal/mol.因此,在丙酮和酸浓度一定的情况下,判断酸辅助单步质子转移为丙酮烯醇化反应的主要途径.
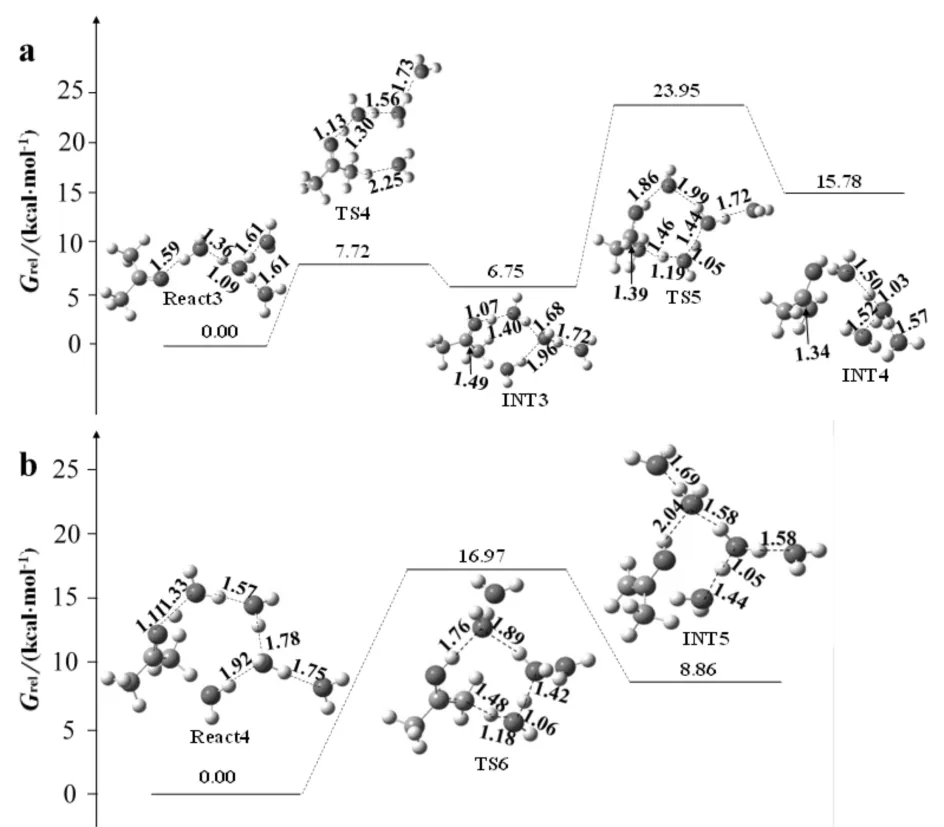
a)分步质子转移反应途径;b) 单步质子转移反应途径图4 酸催化丙酮烯醇化竞争性反应途径的分子结构、相对自由能Grel及部分几何参数Fig.4 Molecular structures, relative free energies Grel and partial geometricparameters for competitive pathways of acid-catalyzed enolization of acetone
反应的第二个阶段是烯醇的碘化反应,反应坐标计算表明:该反应存在3条不同的竞争途径,分图5a描述了碘加成-羟基质子转移反应途径不同阶段的分子结构、相对自由能以及部分关键性参数. 该反应是一个单步历程,始于碘分子进攻烯醇的CC双键. 此途径的自由能能垒值为26.97 kcal/mol.
图5b给出了碘加成-甲基质子转移反应途径及相关参数. 这是一个两步历程.第一步中碘分子首先加成到烯醇的CC双键上,同时夺取另一端甲基上的质子,生成HI及中间体INT6.第二步中,羟基上的质子直接转移到附近的双键碳上.能量计算表明,此途径的活化自由能高达55.82 kal/mol.
图5c描述了碘加成-水辅助羟基质子转移的反应途径及相关参数.反应开始前,碘分子一端通过2个水分子与烯醇羟基形成氢键网络.第一步反应为碘分子对烯醇碳的亲电进攻,由于碘分子的LUMO轨道与烯醇的LUMO轨道形成了有效的σ-π相互作用,碘分子几乎毫无障碍接近CC双键,造成过渡态TS10的相对自由能仅为0.06 kcal/mol.此外,此步中I—I键发生异裂,使烯醇分子带上部分正电,羟基的酸性增加.因此,在后面的质子转移步骤(TS11)中羟基轻易地将其质子转移给临近的水分子.根据能量计算结果,气相状态下质子转移步骤的过渡态(TS11)的相对自由能仅为-5.35 kcal/mol.在溶液中,TS11的相对自由能进一步降至-15.77 kcal/mol,甚至低于中间体INT7,整个反应途径的自由能能垒仅为0.06 kcal/mol,类似于自由基反应.比较图5c与图5a中的分子结构及相应的自由能,可见水分子形成的氢键网络在稳定过渡态方面起着至关重要的作用.

a)碘加成-羟基质子转移途径;b)碘加成-甲基质子转移途径;c)碘加成-水辅助羟基质子转移途径图5 烯醇碘化反应不同途径的分子构型、相对自由能Grel及部分键参数Fig.5 Molecular structures, relative free energiesGrel and partial geometric parameters for various pathways of iodation of enol
比较以上3条反应途径,可见图5c途径的活化自由能远低于其它2条途径的活化自由能,判断碘加成-水辅助羟基质子转移途径为烯醇碘化阶段的主要途径.比较丙酮烯醇化和烯醇碘化2个反应阶段的主要途径,可见烯醇化阶段的酸辅助单步质子转移步骤(图4b)的活化自由能(16.97 kcal/mol)较高,为整个反应的决速步.此步中只有水合质子和丙酮参与了反应,两者的浓度积[H+][A]决定丙酮碘化反应的速率,与速率公式(2)一致.此计算机理较图1能更合理地解释实验观测.
为验证所得计算数据的可靠性,将计算值与实验值进行比较.根据测量实验[14],25 ℃下酸催化丙酮碘化反应的二级速率常数k为0.0286 mmol-1·L·s-1.根据过渡态理论[15],二级速率常数k的计算公式为:
(3)
忽略隧穿效应,利用公式(3)计算该反应的实验活化自由能数值为23.66 kcal/mol.由于反应开始前各反应物相互分离,此活化自由能是处于分离状态的反应物与决速步过渡态的自由能差值.根据这个定义,算得自由状态的丙酮、水合质子的自由能与碘化反应决速步过渡态TS6的差值为25.92 kcal/mol.活化自由能的计算值与实验推测值相符,证明模拟计算可靠.
3 结语
本文运用量子力学方法研究了酸催化丙酮碘化反应的机理.该反应主要有2个子反应构成,即丙酮烯醇化和烯醇碘化反应.丙酮烯醇化存在5条不同的竞争性反应途径,其中酸辅助单步质子转移途径的自由能能垒最低,为16.97 kcal/mol;水分子构成的氢键网络在减小过渡态的环张力方面起重要作用;水合质子需要在氢键网络的协助下才能辅助丙酮将甲基上的质子转移到其羰基氧上.
烯醇碘化反应存在3条竞争性反应途径,其中碘加成-水辅助羟基质子转移途径的自由能能垒最低,仅为0.06 kcal/mol;水分子氢键网络在稳定该反应途径的过渡态方面起至关重要的作用;酸辅助单步质子转移反应是酸催化丙酮反应的决速步.此速决步机理较传统机理更合理地解释了这个反应的速率方程.活化自由能的计算值与实验推测值符合得较好,证明模拟计算及基于此所得机理均为可靠.
