G6PD缺陷对1,4-苯醌致K562细胞DNA甲基化的影响及其机制
2019-06-03章梦莹王博深浦跃朴尹立红
章梦莹,王博深,张 虹,浦跃朴,尹立红,张 娟*
(东南大学公共卫生学院环境医学工程教育部重点实验室,江苏 南京 210009)
苯是一种无色的有机溶剂,是化学工业中常见的原料和试剂,用于制造塑料、油漆、燃料等,是人类日常生活中广泛接触的一种环境污染物[1-2]。苯主要通过皮肤和呼吸道进入体内,在体内经过代谢,形成酚类和醌类物质,进而在骨髓过氧化物酶催化下生成1,4-苯醌(1,4-benzoquinone,1,4-BQ)[3-4]。苯是明确的致癌物[5],大量研究表明苯毒性的潜在机制与苯的代谢产物密切相关,具体毒性机制可能与遗传损伤相关,近年来,发现苯也可引起表现遗传学的改变[6]。
DNA甲基化主要通过DNA甲基转移酶(DNA methyltransferase,DNMTs)进行调控。生物体内的DNMTs 主 要 包 括 : DNMT1、 DNMT3a、 DNMT3b、DNMT3l及DNMT2。研究表明,DNMT1在DNA甲基化进程中维持甲基化状态[7],DNMT3a、DNMT3b在生殖细胞发育和早期胚胎发育过程具备从头甲基化作用[8],DNMT3l通过调控DNMT3a与DNMT3b的表达发挥调节甲基化的作用[9],而DNMT2对DNA甲基化的作用很小[10]。
葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G6PD)缺乏症,俗称“蚕豆病”,是一种红细胞溶血性疾病,全球近4亿人患有该疾病[11]。G6PD是催化戊糖磷酸途径中的第一反应的酶,它以NADPH的形式向所有细胞提供还原能力。NADPH使细胞能够抵抗氧化应激,并保留还原形式的谷胱甘肽。由于红细胞不含线粒体,磷酸戊糖途径是其唯一的NADPH来源,因此,防御氧化损伤取决于G6PD[12-13]。
当细胞处于氧化应激的状态下,G6PD一方面促进NADPH的生成,修复超氧化物引起的氧化损伤,另一方面催化还原性谷胱甘肽的合成,使还原型谷胱甘肽与苯的氧化代谢产物结合排出体外,从而减轻苯造成的氧化损伤。因此,G6PD酶活性对苯所致的氧化损伤具有重要的保护作用,已有研究发现,G6PD酶缺陷的人群对于苯毒性的易感性明显增加[14]。
前期研究表明,G6PD缺陷可增加苯醌染毒K562细胞的氧化损伤,进而引起更高程度的遗传损伤[15]。本研究利用已建立的G6PD缺陷细胞模型,探讨G6PD缺陷对1,4-苯醌致K562细胞株DNA甲基化的影响及其机制,为研究G6PD缺陷人群对苯毒性的易感性提供理论基础。
1 材料与方法
1.1 细胞、试剂和仪器
人慢性髓系白血病细胞,购于中国科学院上海细胞库;伊思柯夫改良培养液、PBS和胎牛血清,购于美国Gibco公司;1,4-BQ购于美国Sigma公司;Realtime PCR扩增试剂盒,购于日本TOYOBO公司;引物购于上海捷瑞生物科技有限公司;实时荧光定量PCR仪,购于美国ABI公司;The MethylFlashTMGlobal DNA Methylation ELISA Easy Kit,购于武汉艾美捷科技有限公司;青霉素和链霉素,购于美国Hyclone公司。
1.2 siRNA慢病毒信息
针对G6PD基因构建RNA干扰慢病毒,包括3个RNA干扰靶点及1个阴性对照。4个慢病毒均进行相应的化学修饰及GFP荧光标记,载体信息为hU6-MCS-Ubiquitin-EGFP-IRES-puromycin。siRNA序列见表1。阴性对照插入序列为TTCTCCCGAACGTGTC ACGT。

表1 siRNA序列表
1.3 体外构建G6PD缺陷细胞株
通过三质粒包装系统将其包装成慢病毒后感染K562细胞构建G6PD缺陷细胞株和阴性对照细胞(G6PD正常表达细胞)。采用Trizol法提取细胞总RNA。所有样品RNA的D(260)/D(280)比值在1.8~2.0。按照试剂盒进行RNA逆转录,逆转录完成后,用Real-time PCR Master Mix进行荧光定量PCR反应,验证G6PD mRNA表达水平,确认细胞构建是否成功。各基因引物序列见表1。反应体系为10 μL(无酶水2.2 μL,SYBR Green qPCR SuperMix 5 μL,Plus溶液1 μL,浓度为10 μmol/L的目的基因上、下游引物各0.4 μL,cDNA 1 μL),qPCR扩增条件为95 ℃、5 min预变性,40个循环扩增(95℃、15 s,60℃、1 min)。每个样品设置3个复孔。采用2-ΔΔCT法计算G6PD mRNA的相对表达水平:ΔCT=CT,目的基因-CT,actin;ΔΔCT=ΔCT,实验组-ΔCT,对照组。
1.4 细胞培养及染毒
将构建成功的G6PD缺陷细胞和G6PD正常表达细胞置于含有10%(体积分数)血清、100 U/mL青霉素和100 μg/mL链霉素的伊思柯夫改良培养液,37℃、CO2体积分数为5%的细胞培养箱内常规培养。待细胞培养至对数生长期后进行分组处理。G6PD缺陷细胞和G6PD正常表达细胞均分别分为4组:①对照组,不进行染毒处理;②加入终浓度为10 μmol/L的1,4-BQ进行染毒;③加入终浓度为20 μmol/L的1,4-BQ进行染毒;④加入终浓度为1.5 mmol/L的GSH和20 μmol/L的1,4-BQ。各组细胞均于染毒12、24、48 h时收获。
1.5 细胞全基因组DNA甲基化检测
1.5.1 DNA提取 将上述步骤处理过的8组细胞收集至15 mL离心管,1 200 r/min离心5 min,弃上清。向离心管中加入2 mL PBS,1 200 r/min离心5 min,弃去上清。收集的细胞加入20 μL Proteinase K溶液,混匀后加入200 μL缓冲液GB,涡旋混匀,70℃放置10 min。待溶液变清亮,瞬时离心以去除管盖内壁的水珠。加人200 μL无水乙醇,充分振荡混匀15 s。将所得溶液转移至吸附柱。向吸附柱加入500 μL缓冲液GD,离心倒掉废液。向吸附柱中加入600 μL漂洗液PW,12 000 r/min离心30 s,倒掉废液。漂洗两遍。将吸附柱置于室温放置数分钟后,将吸附柱转入干净的离心管中,向吸附膜的中间部位悬空滴加50~200 μL洗脱缓冲液TE,室温放置2~5 min,12 000 r/min离心2 min,将溶液收集到离心管中。利用紫外分光光度计检测样品DNA浓度及纯度。
1.5.2 DNA甲基化检测 用纯化的DNA甲基化抗体包被微孔板,制成固相抗体,往包被抗体的微孔中加入样品及标准品。样品孔加入100 ng样品DNA和100 μL结合液。设置6个标准曲线孔,每孔加入100 ng标准品和100 μL结合液,分别含0.1%、0.2%、0.5%、1%、2%、5%的5-甲基胞嘧啶(5-methylcytosine,5-mC)。对照孔加入100 ng试剂盒提供的阴性样品和100 μL结合液。37℃孵育60 min。用150 μL洗涤液洗涤每个孔5次。向每孔添加100 μL显色液,轻轻摇动平板表面5~10 s,在室温下孵育3~4 min。数分钟后,当含5%5-mC的孔颜色变为深蓝色,每孔加入100 μL停止液。轻轻晃动混合溶液并等待1~2 min以使颜色反应完全停止。于酶标仪450 nm处读取每孔吸光度值D(450),时间为2~15 min。计算公式如下:

1.6 实时荧光定量PCR检测DNMTs mRNA表达水平
将经过处理过的8组细胞收集至15 mL离心管,1 200 r/min离心5 min,弃上清。向离心管中加入2 mL PBS,1 200 r/min离心5 min,弃去上清,细胞转移至1.5 mL EP管中,细胞总RNA采用Trizol法提取。NanoPhotometer®spectrophotometer检测RNA纯度。所有样品RNA的D(260)/D(280)比值在1.8~2.0。在逆转录为cDNA后,通过SYBR®Green Real time PCR Master Mix-Plus-试剂盒进行扩增,反应体系为10 μL(无酶水 2.2 μL,SYBR Green qPCR SuperMix 5 μL,Plus溶液1 μL,浓度为10 μmol/L的目的基因上、下游引物各0.4 μL),qPCR扩增条件为95℃、5 min预变性,40个循环扩增(95℃、15 s,60℃、1 min)。每个样品设置3个复孔。采用 2-ΔΔCT法计算mRNA的相对表达水平:ΔCT=CT,目的基因-CT,actin;ΔΔCT=ΔCT,实验组-ΔCT,对照组。引物序列见表2。

表2 基因引物序列
1.7 Western blot检测DNMTs蛋白表达水平
利用蛋白酶抑制剂和磷酸酶抑制剂冰上裂解细胞液,12 000 r/min离心8 min,留取上清。采用BCA方法检测蛋白浓度。选用SDS-PAGE凝胶电泳,转移至PVDF膜,5%脱脂牛奶封闭2 h,加入1∶1 000稀释的一抗(ACTIN、DNMT1和DNMT3a)在4℃下孵育过夜。第二日加入1∶5 000稀释的二抗(HRP标记山羊抗兔二抗)室温孵育1 h,TBST洗涤6次,用显色液曝光,化学发光成像系统捕捉保存,并对蛋白条带的相对表达水平进行分析。
1.8 统计学方法
采用SPSS 17.0软件对结果进行分析。同种细胞的不同剂量组间全基因组甲基化相对水平、DNMTs mRNA表达水平及DNMT1和DNMT3a蛋白表达水平采用单因素方差分析。同一剂量组的G6PD缺陷细胞和G6PD正常细胞的全基因组甲基化相对水平、DNMTs mRNA表达水平及DNMT1和DNMT3a蛋白表达水平的比较采用两独立样本t检验。同一剂量的1,4-BQ不同处理时间组间的DNA甲基化相对水平的比较采用单因素方差分析。以α=0.05为检验水准。
2 结果
2.1 G6PD缺陷K562细胞的鉴定
采用荧光定量PCR方法检测K562细胞中G6PD mRNA表达水平,结果见图1,发现G6PD缺陷细胞G6PD mRNA表达水平显著低于对照K562细胞(P<0.05),说明G6PD缺陷的K562细胞株构建成功。
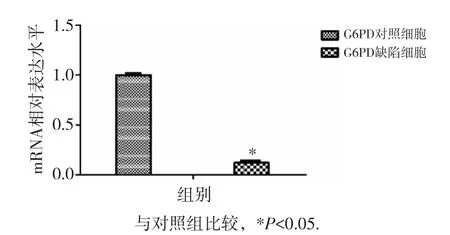
图1 荧光定量PCR方法检测K562细胞中G6PD mRNA表达水平
2.2 1,4-BQ染毒后K562细胞基因组DNA甲基化水平
1,4-BQ染毒K562细胞24 h时基因组DNA甲基化水平见图2A,可见在未染毒及相同浓度1,4-BQ染毒时,G6PD缺陷细胞基因组DNA甲基化水平均较G6PD正常表达细胞升高(P<0.05)。在1,4-BQ浓度为20 μmol/L时,G6PD正常表达细胞和G6PD缺陷细胞基因组DNA甲基化水平均较相应的未染毒细胞升高(P<0.05)。1,4-BQ染毒不同时间后K562细胞全基因组DNA甲基化相对水平见图2B、C,可见经10 μmol/L 1,4-BQ染毒的48 h内,以及20 μmol/L 1,4-BQ染毒的24 h内,G6PD缺陷细胞的DNA甲基化相对水平均高于G6PD正常表达细胞(P<0.05)。另外,经过20 μmol/L 1,4-BQ染毒48 h时,两种细胞的甲基化相对水平均有所下降,此时G6PD缺陷细胞的全基因组DNA甲基化相对水平低于G6PD正常表达细胞(P<0.05)。
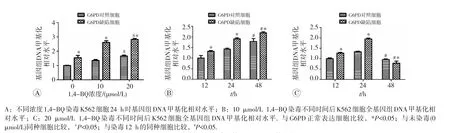
图2 1,4-BQ染毒K562细胞后基因组DNA甲基化水平
2.3 DNMTs mRNA的表达水平
实时荧光定量PCR检测结果见图3,在0、10、20 μmol/L共3种不同剂量1,4-BQ染毒下,除10 μmol/L 1,4-BQ染毒时的DNMT3b mRNA,其他各组中G6PD缺陷细胞DNMT1、DNMT3a及DNMT3b mRNA的表达水平均高于G6PD正常表达细胞(P<0.05)。在1,4-BQ浓度为20 μmol/L时,G6PD正常表达细胞和G6PD缺陷细胞DNMT1、DNMT3a及DNMT3b mRNA的表达水平均较相应的未染毒细胞升高(P<0.05)。

图3 实时荧光定量PCR检测K562细胞DNMTs mRNA的表达
2.4 DNMT1、DNMT3a蛋白表达水平
Western blot检测结果见图4,在0、10、20 μmol/L共3种不同剂量1,4-BQ染毒下,G6PD缺陷细胞的DNMT1和DNMT3a蛋白表达水平均高于G6PD正常表达细胞(P<0.05)。另外,在1,4-BQ浓度为20 μmol/L时,G6PD正常表达细胞和G6PD缺陷细胞DNMT1和DNMT3a蛋白的表达水平均较相应的未染毒细胞升高(P<0.05)。
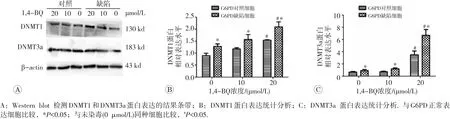
图4 Western blot检测K562细胞DNMT1、DNMT3a蛋白的表达
2.5 GSH干预对K562细胞全基因组DNA甲基化的影响
为验证GSH的作用,在20 μmol/L 1,4-BQ染毒时,同时在培养液中加入GSH进行干预,检测细胞全基因组DNA甲基化水平,结果图5。20 μmol/L 1,4-BQ染毒后,G6PD缺陷细胞基因组DNA甲基化水平较G6PD正常表达细胞升高(P<0.05)。加入GSH后,两组细胞基因组DNA甲基化水平差异无统计学意义(P>0.05)。
2.6 GSH干预对K562细胞DNMTs mRNA表达水平的影响
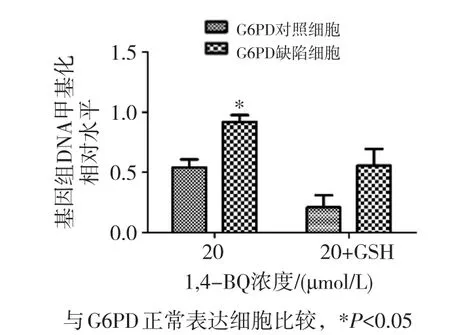
图5 GSH干预对K562细胞全基因组DNA甲基化的影响
实时荧光定量PCR检测结果见图6,20 μmol/L的1,4-BQ染毒后,G6PD缺陷细胞DNMT1、DNMT3a及DNMT3b mRNA表达水平均较G6PD正常表达细胞升高(P<0.05)。加入GSH后,两组细胞DNMT1、DNMT3a及DNMT3b mRNA表达水平差异无统计学意义(P>0.05)。
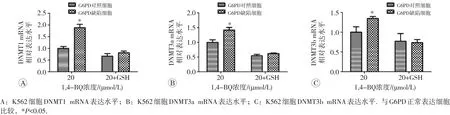
图6 实时荧光定量PCR检测GSH干预对DNMTs mRNA表达水平的影响
2.7 GSH干预对K562细胞DNMT1、DNMT3a蛋白表达水平的影响
Western blot检测结果见图 7,20 μmol/L1,4-BQ染毒后,G6PD缺陷细胞DNMT1及DNMT3a蛋白表达水平均较G6PD正常表达细胞升高(P<0.05)。加入GSH后,两组细胞DNMT1及DNMT3a蛋白表达水平差异无统计学意义(P>0.05)。
3 讨论
G6PD是磷酸戊糖途径的限速酶,G6PD缺乏使GSH生成量减少,影响以GSH为中心的抗氧化系统,从而加重机体自身氧化损伤[16]。研究发现,G6PD缺陷细胞的甲基化相对水平高于对照组,原因可能是G6PD缺陷导致GSH减少,使细胞更容易受到外界自由基攻击,使细胞甲基化相对水平进一步上升。
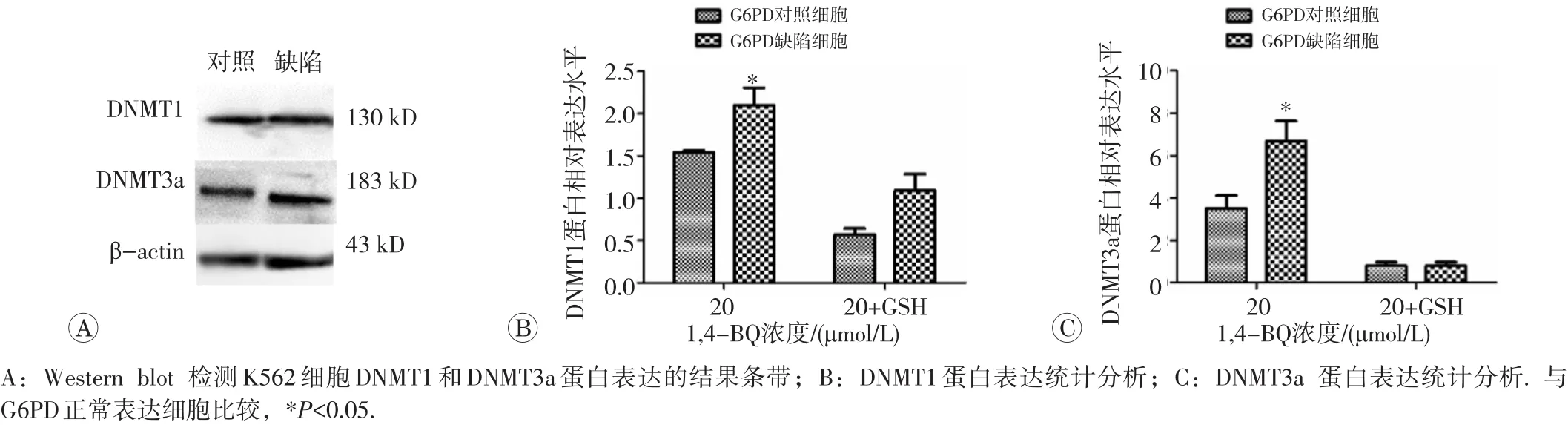
图7 Western blot检测GSH干预对K562细胞DNMT1、DNMT3a蛋白表达水平的影响
当前,国家化工产业以苯为原材料的生产量大,苯作业工人众多,但由于苯致癌的具体机制仍不清楚,所以寻找有效的生物标志一直是防控职业苯中毒的研究热点[17]。在我国,苯作业工人健康保护和体检筛查首先以外周血象为主,主要依赖于外周血白细胞水平,但目前已有研究发现,相比于苯引起的血象变化,表观遗传具有更为直接和早期的表现[18]。
通过全基因组甲基化分析,发现采用10 μmol/L 1,4-BQ染毒K562细胞时,细胞的甲基化水平在48 h内持续升高(P<0.05)。两种细胞被苯醌处理后,甲基化相对水平均出现了升高。推测细胞因被苯醌处理引发氧化应激,促进自身自由基生成量增大,氧化损伤加重,甲基转移酶的活性改变,进而改变全基因组DNA甲基化的水平。K562细胞在经过20 μmol/L 1,4-BQ染毒后的12~24 h内,甲基化水平相对升高,但在24~48 h内又会有所降低。此现象原因可能是细胞经苯醌染毒24 h后,细胞存活量减少,导致甲基化水平降低。经过1,4-BQ染毒后,G6PD缺陷细胞和G6PD正常细胞的甲基化相对水平差异显著增大(P<0.05)。这是由于G6PD缺陷细胞受到1,4-BQ毒性作用时,G6PD缺陷细胞不能产生足够的抗氧化物质,无法代谢1,4-BQ,使自身更容易受到1,4-BQ的损伤。
由于DNA甲基化相对水平受到DNA甲基转移酶的调控,故本研究进一步检测了G6PD缺陷细胞与正常表达细胞的DNMTs表达水平。研究发现,G6PD缺陷细胞的DNMT1、DNMT3a、DNMT3b mRNA表达量均高于G6PD正常细胞。1,4-BQ处理后,G6PD缺陷细胞与G6PD正常细胞的DNMTs表达水平的差异显著增大。进一步检测了DNMT1和DNMT3a蛋白的表达水平。结果显示,相比于G6PD正常细胞,G6PD缺陷细胞的DNMT1、DNMT3a蛋白表达水平更高(P<0.05)。推测G6PD缺陷细胞更容易受到氧化损伤,引起自由基增多,导致甲基化转移酶表达升高,进而甲基化相对水平更高。
为了验证GSH生成不足是导致G6PD缺陷细胞的甲基化相对水平升高重要原因的猜想,本研究在20 μmol/L苯醌染毒细胞的基础上加入GSH进行干预。通过在培养液中加入GSH,G6PD缺陷细胞与对照细胞的DNA甲基化相对水平差异无统计学意义(P>0.05)。表明GSH可以消除G6PD缺陷所致的全基因组DNA甲基化相对水平及DNMTs mRNA表达水平的改变。
同样在1,4-BQ影响下,G6PD缺陷细胞的甲基化相对水平会明显高于正常细胞,由此可见,G6PD缺陷可能以通过抑制GSH合成的方式增加氧化应激水平,最终导致细胞DNMTs生成量的增多以及全基因组DNA甲基化程度的升高。
