Halomonas sp.QHL1四氢嘧啶合成基因簇ectABC与上游调控序列的克隆及其功能分析※
2019-05-08朱德锐
石 晴,朱德锐
(青海大学医学院 基础医学研究中心 西宁 810016)
四氢嘧啶[Ectoine:(4S)-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid]是一类嗜盐或耐盐菌胞内合成的主要相容溶质,是用于抵抗高盐环境的一种重要物质[1]。其结构属氨基酸环化衍生物或部分氢化嘧啶衍生物[2],具有分子手性,化学方法难以合成[3]。在生化特性方面,Ectoine具有惰性、胞内高耐受性、热稳定性和水溶性等特点[2],影响细胞在逆境环境中的渗透压调节功能,可提高细胞在高盐、干旱和低温等恶劣环境中的适应性。Ectoine作为微生物重要的次级代谢产物,已广泛应用于生物医学领域[4-7]。
近年来,Ectoine的生物合成途径从基因水平、酶水平和调控水平等方面已有较为深入的研究。用ectA、ectB和ectC3个基因分别编码的L-2,4-二氨基丁酸乙酰转移酶(Ect A)、L-2,4-二氨基丁酸转氨酶(Ect B)和Ectoine合成酶(Ect C)调控Ectoine的生物合成[8]:首先,以L-天冬氨酸-β半醛(ASA)为底物在Ect B催化作用下生成L-2,4-二氨基丁酸(DABA);其次,Ect A将DABA乙酰化生成N-乙酰-L-2,4-二氨基丁酸(ADABA);最后,将ADABA催化生成四氢嘧啶。基于此,克隆及异源表达Ectoine合成基因簇及其上游调控序列,用于阐明在盐激条件下何种调节蛋白或因子参与Ectoine合成基因簇ectABC操纵子的启动应答的分子机制。本研究以Halomonassp.QHL1为模式菌株,克隆ectABC基因簇及其上游调控序列,并对其在E.coilBL21中的蛋白表达及耐盐能力进行检测。
1 材料与方法
1.1 实验材料
Halomonassp.QHL1菌株分离自青海湖水体;菌株E.coilDH5α和E.coilBL21(DE3)由青海大学医学院基础医学研究中心提供;细菌基因组DNA提取试剂盒、质粒小提试剂盒(离心柱)和GeneGreen核酸染料购自北京天根生化科技有限公司;克隆载体pMD18-T、表达载体pColdⅠDNA、T4 DNA连接酶和限制性内切酶购自大连TaKaRa公司;DL 5000 Maker购自广东东盛生物科技有限公司;AxyPrep DNA凝胶回收试剂盒购自杭州爱思进生物技术有限公司。引物合成及DNA测序由南京金斯瑞生物科技有限公司完成。
OSM液体培养基(g/L):超纯水内加MgSO4·7H2O 20 g、KCl 2 g、Na3C6H5O7·2H2O 3 g、无水CaCl20.2 g、Yeast Extract 2 g、Tryptone 10 g、C6H12O65 g、NaCl 29.25 g,定容至1 L,调节pH值至7.5,灭菌(120℃)20 min。
LB液体培养基(g/L):超纯水内加Tryptone 10 g、Yeast Extract 5 g、NaCl 10 g,定容至1 L,灭菌(120℃)20 min。
LA固体培养基:每升LB液体培养基中加入琼脂粉15 g,灭菌(120℃)20 min,待培养基冷却后,按照100 mL培养基加100 μL氨苄青霉素(终浓度为50mg/mL)的原则配制。
1.2 实验方法
1.2.1Halomonassp.QHL1基因组DNA的提取
将Halomonassp.QHL1菌株接种于OSM液体培养基中培养(37℃,180r/min,过夜)。DNA提取参照细菌基因组DNA提取试剂盒说明书进行。
1.2.2 QHL1ectABC基因簇及其上游启动子序列的克隆
本课题组[9]前期已对Halomonassp.QHL1全基因组序列做生物信息学分析,获得ectABC及其上游调控序列,利用Primer 5.0软件设计出特异性引物(表1)。以Halomonassp.QHL1全基因组DNA为模板,将F1/R1、F2/R2和F3/R3交叉配对进行PCR扩增。PCR反应体系(50μL):DNA模板2.5 μL;引物各1 μL;dNTP 4 μL;LA Taq酶1 μL;10×buffer(含Mg2+)5 μL;灭菌双蒸水35.5 μL。反应条件:94 ℃预变性3 min;94 ℃变性30 s,55 ℃退火30 s,72 ℃延伸3 min,共35个循环;72 ℃下延伸10 min。行1%琼脂糖凝胶电泳检测。用凝胶回收试剂盒纯化回收PCR产物,与pMD18-T载体TA互补连接后转化至E.coilDH5α,筛选阳性质粒做酶切验证并测序。
1.2.3 重组表达载体的构建及验证
将阳性质粒pMD18-T-promoter+ectABC和表达载体pColdⅠDNA双酶切(37℃)4 h后,纯化回收酶切产物,经T4 DNA连接酶连接(过夜,16℃),转化至E.coilBL21(DE3)。根据重组表达载体pColdⅠDNA-promoter+ectABC的酶切位点进行酶切验证,使用限制性内切酶BamHⅠ/PstⅠ做双酶切、BamHⅠ做单酶切,挑取阳性重组子测序。
1.2.4 重组菌株的诱导表达与鉴定
将重组菌株E.coilBL21/pColdⅠDNA-promoter+ectABC以IPTG时间梯度方法诱导:用LB培养基(50μg/mL Amp)诱导培养重组菌株48 h(15℃,180r/min,0.1 mmol/L IPTG),间隔12 h取样。离心收集诱导培养后的菌体沉淀,加入1×PBS将菌体冲悬,在冰上超声裂解至液体清亮。离心(12000r/min)5 min,收集上清液,用SDS-PAGE检测表达产物,以未诱导的E.coilBL21和pColdⅠDNA空载作阴性对照。
1.2.5 重组菌株的盐耐受实验
将E.coilBL21菌株、E.coilBL21/pColdⅠDNA空载以及E.coilBL21/pColdⅠDNA-promoter+ectABC重组菌株使用LB液体培养基活化(37℃,培养至光密度值OD600约为0.6~0.8),分别接种至不同NaCl浓度的LB培养基中,同一梯度设3个平行组,培养(37℃)12 h后采用紫外分光光度计测定OD600值,求均值及标准差。
表1本实验中的引物序列

Table 1Primer sequences used in this study
2 结果与分析
2.1 ectABC基因簇及其上游启动子序列的克隆
以QHL1的基因组DNA为模板,用F1和R3这一对引物特异性扩增得到四氢嘧啶合成基因簇ectABC及其上游启动子序列,经电泳分析可知,片段大小与预期结果一致(图1),约为3 335 bp。纯化回收PCR产物,与pMD18-T载体连接,构建重组克隆载体pMD18-T-promoter+ectABC,并转化至大肠杆菌DH5α构建阳性质粒。经酶切(图2)及测序验证,显示阳性质粒构建成功。
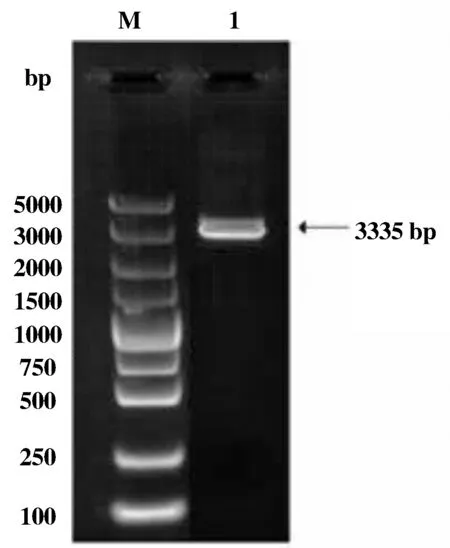
图1Halomonassp.QHL1的ectABC及其上游启动子基因序列琼脂糖电泳分析图
Figure1The analysis ofectABCand its upstream regulation sequence fromHalomonassp.QHL1by agarose gel electrophoresis

图2 重组质粒pMD18-T-promoter+ectABC的酶切鉴定图
2.2 重组表达质粒的构建
将阳性质粒pMD18-T-promoter+ectABC和表达载体pCold Ⅰ DNA的酶切产物连接,获得重组表达载体pCold Ⅰ DNA-promoter+ectABC,转化至E.coilBL21(DE3)。重组质粒pCold Ⅰ DNA-promoter+ectABC经BamH Ⅰ和PstⅠ双酶切后,目的片段约3 335 bp,与预期大小一致(图3)。经测序验证,显示重组质粒构建成功。
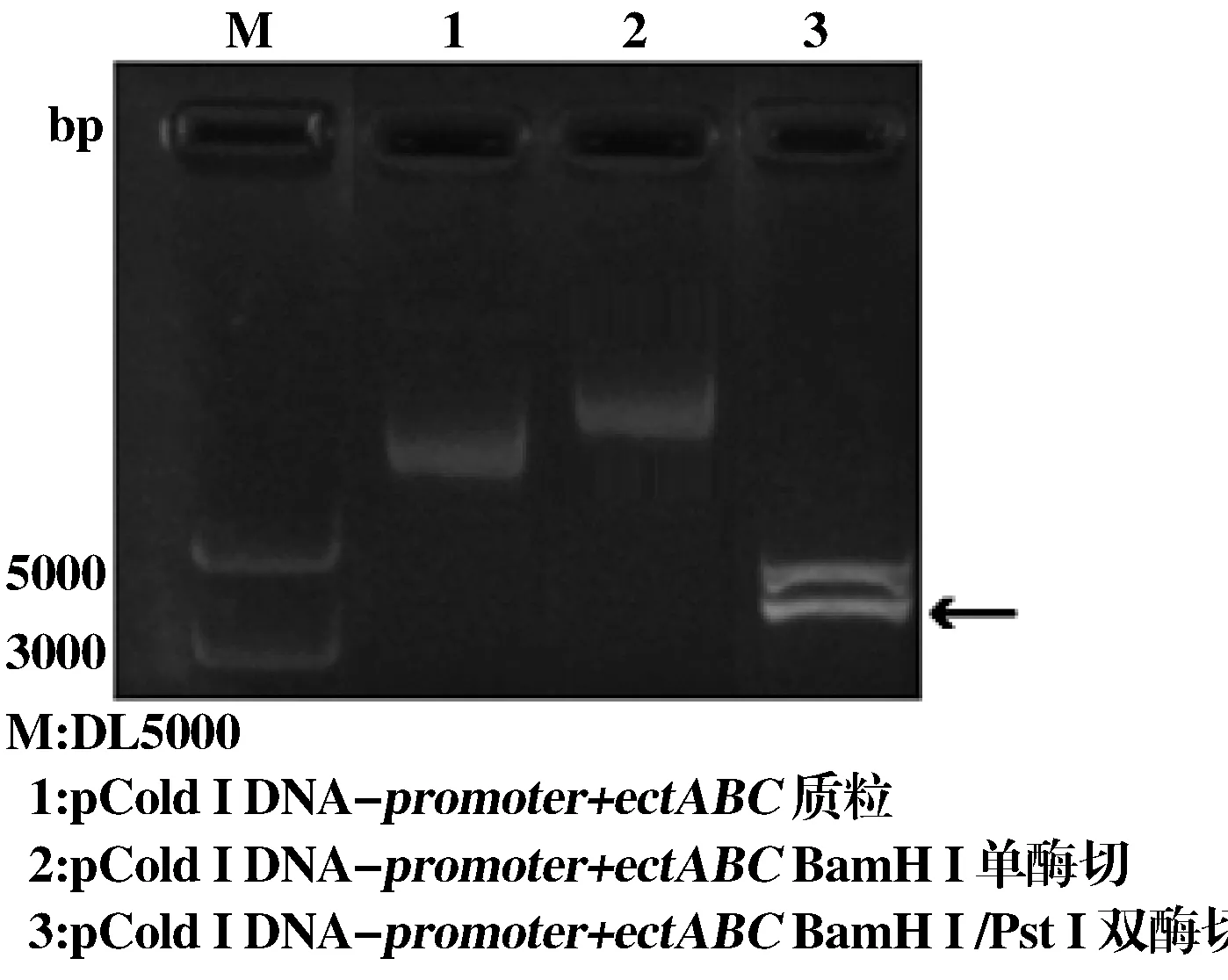
图3重组质粒pCold Ⅰ DNA-promoter+ectABC的酶切鉴定图
Figure3Identification of enzyme digestion of recombinant plasmid pCold Ⅰ DNA-promoter+ectABC
2.3 QHL1 ectABC基因簇及其上游启动子序列的异源表达
将重组表达菌株E.coilBL21/pCold Ⅰ DNA-promoter+ectABC用IPTG进行诱导,对比发现IPTG浓度为0.1 mM、诱导时间为12 h时,目的蛋白异源表达效果最佳。SDS-PAGE结果显示(图4):ectA、ectB和ectC分子量大小分别为:21.2、46.4、14.7 kDa,与预测结果一致,表明promoter+ectABC基因簇在E.coliBL21中能实现异源共表达。
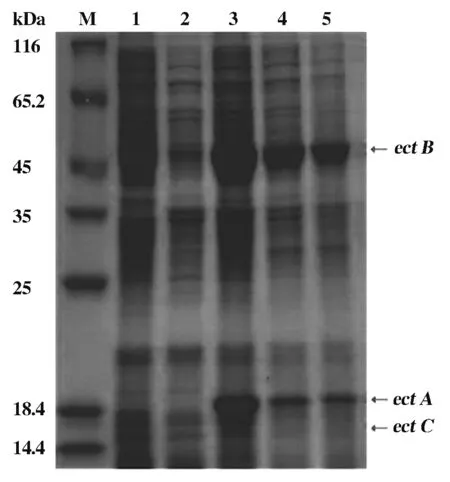
M为蛋白Marker;1和2分别为未诱导的E.coilBL21和E.coilBL21/pCold Ⅰ DNA空载体;3到5分别为诱导12、24和48 h的重组菌株E.coilBL21/pColdⅠDNA-promoter+ectABC(0.1mM IPTG)
图4SDS-PAGE分析promoter+ectABC基因簇在E.coilBL21中的异源表达
Figure4Heterologous expression of thepromoter+ectABCgene cluster in theE.coilBL21by SDS-PAGE analysis
2.4 重组菌株E.coil BL21/pColdⅠDNA-promoter+ectABC的耐盐能力检测
配制不同盐浓度梯度(0~1.3mol/L NaCl)的LB培养基,检测自带启动子序列的ectABC基因簇的耐盐能力。结果如图5显示:E.coilBL21耐盐浓度为0.8 mol/L;而E.coilBL21/pCold Ⅰ DNA-promoter+ectABC耐盐浓度达到1.3 mol/L,且生长量明显高于对照菌株。与原始菌株相比,重组菌株耐盐性明显提高,能够抵抗一定水平的渗透压冲击。

图5重组菌株与对照菌株盐耐受实验图
Figure5Salt tolerance of the nativeE.coilandE.coilengineered
3 讨论
盐度是影响微观和宏观微生物群落组成的主要环境因素之一[10]。随着盐度的增加,多细胞生物的物种丰富度下降,在最高盐度时,动、植物消失,仅剩下古菌和细菌、真菌及原生生物等微生物[11]。研究发现,相容性溶质在应对高渗透压方面起着重要作用[12],这些相容性溶质主要包括糖(如蔗糖和海藻糖)、多元醇(如甘油、山梨糖醇、甘露醇、α-葡萄糖基甘油、甘露糖基甘油和甘露糖基甘油酰胺)、甜菜碱(如甘氨酸甜菜碱和衍生物)和氨基酸及其衍生物(如脯氨酸、谷氨酸、谷氨酰胺、丙氨酸、四氢嘧啶和羟基四氢嘧啶)等[13]。Ectoine是一种水结合两性离子氨基酸衍生物,最早由Galinski等人在ctothiorhodospirahalochloris中发现[14]。Ectoine在各类中度嗜盐菌的细胞中广泛积聚,是嗜盐微生物在高盐胁迫下维持渗透压平衡的一类主要相容性溶质[15]。当高渗冲击时,微生物在胞内大量积聚Ectoine以平衡高渗透压;当渗透压降低时,微生物则向胞外释放Ectoine[16-17]。此外,Ectoine对细胞、蛋白质和核酸等还能起到抗冷冻、干旱、高温、高盐、辐射等作用[18]。
目前,已报道参与Ectoine生物合成的基因类型有ectABC、ectABCD、ectABC-ask和ectABCD-ask[19-20],其生物合成相关基因已从多种微生物中克隆获得,如唐娜等[21]通过SEFA-PCR步移方法从盐单胞菌属H.sociaNY-011T中克隆获得ectABC和ectD基因,实现了E.coil异源表达;张慧艳等[22]从易变链霉菌TRM 45540中克隆了ectABC基因并实现了E.coil异源表达,且携带ectABC基因工程菌的耐盐能力显著提高;高波等[23]从密旋链霉菌Act12中克隆了基因簇ectABC并在E.coil中成功异源表达,但重组工程菌株的盐耐受能力无明显改善。大肠杆菌因其遗传背景清晰,遗传操作技术熟练,与外源基因具有良好相容性的系统等特点[24],被作为广泛使用的工业菌株,用于构建产生外蛋白的宿主生物。本研究中,模式菌株QHL1分类地位为盐单胞菌属,通过BLST比对分析发现Ectoine生物合成基因类型为ectABC。以QHL1基因组序列为模板,设计ectABC及其上游启动子序列的引物,克隆获得Halomonassp.QHL1四氢嘧啶生物合成操纵子序列(3335bp)并在E.coilBL21中实现异源表达,且重组菌株(E.coilBL21/pCold Ⅰ DNA-promoter+ectABC)的盐耐受能力明显提高。
