Central role of Yes-associated protein and WW-domain-containing transcriptional co-activator with PDZ-binding motif in pancreatic cancer development
2019-05-08EnriqueRozengurtGuidoEibl
Enrique Rozengurt, Guido Eibl
Abstrac t Pancreatic ductal adenocarcinoma (PDAC) remains a deadly disease with no efficacious treatment options. PDAC incidence is projected to increase, which may be caused at least partially by the obesity epidemic. Significantly enhanced efforts to prevent or intercept this cancer are clearly warranted. Oncogenic KRAS mutations are recognized initiating events in PDAC development, however, they are not entirely sufficient for the development of fully invasive PDAC.Additional genetic alterations and/or environmental, nutritional, and metabolic signals, as present in obesity, type-2 diabetes mellitus, and inflammation, are required for full PDAC formation. We hypothesize that oncogenic KRAS increases the intensity and duration of the growth-promoting signaling network.Recent exciting studies from different laboratories indicate that the activity of the transcriptional co-activators Yes-associated protein (YAP) and WW-domaincontaining transcriptional co-activator with PDZ-binding motif (TAZ) play a critical role in the promotion and maintenance of PDAC operating as key downstream target of KRAS signaling. While initially thought to be primarily an effector of the tumor-suppressive Hippo pathway, more recent studies revealed that YAP/TAZ subcellular localization and co-transcriptional activity is regulated by multiple upstream signals. Overall, YAP has emerged as a central node of transcriptional convergence in growth-promoting signaling in PDAC cells. Indeed, YAP expression is an independent unfavorable prognostic marker for overall survival of PDAC. In what follows, we will review studies implicating YAP/TAZ in pancreatic cancer development and consider different approaches to target these transcriptional regulators.
Key words: Pancreatic cancer; Yes-associated protein and WW-domain-containing transcriptional co-activator with PDZ-binding motif; Oncogenic Kras; Obesity; Signaling network and loops
INTRODUCTION
Despite advances in our understand ing of genetics and basic biology, imaging,surgical treatments, and adjuvant therapy, p ancreatic ductal ad enocarcinoma(PDAC), which represents 90% of all pancreatic cancers, is a disease with dismal prognosis with an overall 5-year survival rate of only about 7%[1]. The incidence in the general population is estimated to be 8 cases per 100000 person-years, and the worldwide mortality about 7 deaths per 100000 person-years[2,3]. PDAC is already the 3rdleading cause of cancer-related mortalities in the United States[4]. Indeed, deaths due to PDAC are predicted to increase markedly. Indeed, the disease is expected to become the 2ndleading cause of cancer-related mortality in the United States in the next few years[5]. Given that only a minority of patients diagnosed with PDAC are eligible for surgical intervention, the research is gradually shifting to identify novel approaches in early diagnosis, prevention and interception, a novel concept, which attempts stopping transformed cells from progressing to frank cancer[6-10]. The elucidation of the molecular mechanisms of risk-factor associated PDAC promotion will be of paramount importance to facilitate the discovery of novel targets and agents for prevention and identify robust biomarkers to stratify patients for selective and individualized therapeutics.
KRAS MUTATIONS AND PDAC
Oncogenic KRAS mutations w ere first reported to be associated w ith PDAC more than 30 years ago[11,12]. Although the genetic landscape of PDAC is complex, since the initial reports extensive research in both humans and mice have substantiated the critical significance of KRAS mutations in the early stages of PDAC. In fact, many stud ies have confirmed that over 90% of PDAC harbors KRAS mutations[13,14]and KRAS signaling is one of the core signaling pathw ays in human PDAC[13]. Most KRAS mutations in PDAC are found at p osition G12, of w hich the single amino acid replacement G12D is the most predominant[15]. Mutations at position G13 or Q61 have been detected at low er frequency, 21% or 28%, respectively[15]. Using d eep w hole exome sequencing an integrated genomic characterization of PDAC revealed several different KRAS mutations in a subset of tumors, with some PDACs showing biallelic mutations[16]. Mechanistically, mutations at position G12 w ith a single amino acid substitution induce conformational changes that interfere w ith the intrinsic GTPase activity of KRAS and prevent the interactions between KRAS and GTPase-activating proteins (GAPs), w hich stimulate the conversion of KRAS-GTP (active state) to KRASGDP (inactive state), thereby end ing KRAS activation. In this manner, the KRAS mutations lead to its p rolonged activation and consequently to the p ersistent stimulation of d ow nstream signaling effectors[15,17]. It is becoming clear that different mutations of G12 lead to different conformational states that differ in their affinity for interacting effectors[18]. Although mutations in KRAS is an early and essential step in PDAC, it is insufficient to stimulate development of frank, invasive PDAC. Activation of other pathways by additional mutations (e.g., in tumor suppressor genes, including p53, p 16 and SMAD4) or environmental stimuli, includ ing obesity and metabolic syndrome are required for the promotion of invasive PDAC[19-24].
In ad d ition, the “efficacy” of oncogenic KRAS to initiate and promote PDAC is influenced and modulated by the presence of common susceptibility genes. Recent genome-w ide association studies (GWAS) of PDACs in p op ulations of European ancestry have id entified ad d itional common pancreatic cancer risk loci carrying p ancreatic cancer risk signals, includ ing NR5A2, PDX1, AB0, NOC2L, HNF1B,GRP[25-28]. Moreover, an elegant stud y d emonstrated that variations in oncogenic dosage have a critical role in PDAC biology and phenotypic diversification[29], w ith the highest oncogenic Kras levels und erlying aggressive und ifferentiated phenotypes.Activation of other pro-oncogenic p athw ays, includ ing Myc, Yap1 or Nfkb2, w hich collaborate w ith heterozygous mutant Kras in d riving tumorigenesis have been show n to have a low er metastatic potential[29]. It seems that evolutionary constraints direct oncogenic d osage gain and variation along defined routes to drive the early progression of PDAC and shape its dow nstream biology[29]. Integrated genomic and global gene expression analyses have classified human pancreatic cancers into several d istinct subtypes that may d ictate and p red ict clinical outcomes and therap eutic resp onses. Collison and colleagues d efined three subtyp es: classical, quasi mesenchymal, and exocrine-like[30], w hile Bailey et al[31]classified four subtyp es:squamous, p ancreatic p rogenitor, immunogenic, and aberrantly d ifferentiated end ocrine exocrine (ADEX). By separating tumor cells and stromal components,Moffitt and colleagues identified tw o stromal subtypes: normal and activated, and tw o tumor-sp ecific subtyp es: classical and basal-like[32]. Using w hole genome sequencing and cop y number variation analysis Wad dell et al[33]categorized PDAC into four subclasses based on p atterns of structural variation (variation in chromosomal structure): stable, locally rearranged, scattered, and unstable. Taken together, these large genomic efforts clearly d emonstrate that pancreatic cancer is a genetically complex and heterogeneous disease, which has significant implications in prognosis and therapeutic response, and classifying pancreatic cancers into subtypes may assist and pave the way to more efficacious personalized treatment strategies.
PROGRESSION MODEL OF PDAC
It is estimated that PDACs develop over many years from non-invasive precursor lesions. The non-cystic lesion is called pancreatic intraepithelial neoplasia (PanIN) and is usually diagnosed in histological preparation of tissue removed during surgery or in biopsy specimens[34-37]. These Pan INs progress from early Pan IN-1 lesions to advanced PanIN-3 (carcinoma in situ) and finally to frank invasive PDAC. Besides this classical view of gradual step-wise Pan IN progression and PDAC formation, in at least a subset of PDACs there seem to be catastrop hic genetic events (e.g.,chromothripsis) necessary for the transition from preinvasive to invasive PDAC(punctuated equilibrium)[38-41]. The pathological characteristics of cystic precursor lesions, including intraductal papillary mucinous neoplasm (IPMN) have been recently reviewed elsewhere[42]. Most low-grade Pan IN lesions contain oncogenic KRAS mutations[43]. This finding provided further evidence in support of the step-wise carcinogenesis model, in w hich KRAS mutations are envisioned as initiating events[15,44,45].
Genetically engineered mouse mod els of PDAC have corroborated this paradigm[46-49]. In the KC model, mutated Kras is expressed from its endogenous locus(by crossing LSL-KrasG12D mice with PDX-1-Cre or p48-Cre mice, i.e., KC model)[48-50].This KC mouse model shares similar histopathologic and genetic features to the human disease including the development and progression of Pan INs[46]. In addition to the role of oncogenic KRAS in the initiation of PDAC, Kras mutations have also been shown to be important for PDAC maintenance[51,52]. In line with the notion that mutated Kras is necessary but not fully sufficient for the development of invasive PDAC, only few animals (5%-10%) in the KC model (without additional genetic alterations) develop frank PDAC very late (usually after 9 mo)[46]. Cell senescence has been proposed as a barrier to the malignant progression of tumors[53]. The formation of PDAC can be greatly accelerated by the presence of another mutation (e.g., Trp53)[47,54].
Besid es ad d itional genetic mutations, sev eral stud ies hav e convincingly d emonstrated that environmental, nutritional, and metabolic factors, includ ing obesity, type-2 diabetes mellitus (T2DM) and inflammation efficiently promote PDAC formation[55-59]. This notion is substantiated by several preclinical studies. Expression of physiologic levels of oncogenic Kras in murine models efficiently transformed only a small percentage of cells[60]. KRAS dow nstream signaling molecules, including the ERKs w ere not activated w hen oncogenic Kras w as expressed from its endogenous locus[61]. Accordingly, cell culture studies have shown that incubating PDAC cells in a serum-free medium failed to display activated ERK d esp ite the presence of KRAS activating mutations in these cells. How ever, ERK activation could be induced by adding growth factors to the culture medium[62-64]. In mouse models, oncogenic Kras in ad ult mice w as insufficient to ind uce PDAC, w hile concomitant ind uction of p ancreatic inflammation (e.g., by ad ministration of the cholecystokinin analog cerulein) stimulated the formation of Pan INs and cancers[65]. Our ow n stud ies have clearly demonstrated that an obesogenic diet accelerated early PanIN progression and PDAC development in KC mice, which was associated w ith metabolic disturbances(e.g., hyperinsulinemia), increased p ancreatic inflammation, and d esmoplasia[55,56].Taken together, the current evid ence indicates that oncogenic Kras is ind isp ensable but not sufficient to ind uce malignant p ancreatic cells. Ad d itional genetic or environmental factors (obesity, T2DM, inflammation) are required to elevate KRAS activity[52]and/or stimulate ad d itional signaling p athw ays to p romote PDAC formation[66].
Recent elegant gene-environment interaction stud ies have demonstrated that the increased risk of developing PDAC by environmental stimuli and conditions may be influenced by the presence of common genetic variations. A GWAS data analysis has found that genetic variations in inflammatory responses and insulin resistance may affect the risk of obesity- and diabetes-related pancreatic cancer[67]. It is apparent that a detailed und erstand ing of the gene-regulatory netw orks that integrate signaling by KRAS and cooperating pathw ays to drive an oncogenic program in pancreatic cancer is of fund amental imp ortance to d esign novel strategies to target this aggressive disease. Recent exciting studies from different laboratories indicate that the activity of the transcrip tional regulators yes-associated Protein (YAP) and WW-d omaincontaining transcriptional co-activator with PDZ-binding motif (TAZ) play a critical role in the p romotion and maintenance of PDAC. In w hat follow s, w e w ill review stud ies imp licating YAP/TAZ in p ancreatic cancer d evelop ment and consid er p ossible ap p roaches to target these transcrip tional regulators w ith emp hasis in repurposing drugs that are currently in clinical use.
YAP/TAZ IN PANCREATIC CANCER
The Hippo pathway
The highly conserved Hip p o pathw ay, originally id entified and characterized as potent grow th-suppressive pathw ay in Drosophila[68], is a key regulatory mechanism in development, organ-size, tissue regeneration and tumorigenesis[68,69]. Canonical Hippo signals are transmitted via the serine/threonine kinases mammalian Ste20-like kinases 1/2 (Mst1/2), in complex with the scaffold protein salvador homolog 1 (Sav1),phosphorylate and activate large tumor suppressor 1/2 (Lats1/2), in complex w ith its regulatory protein Mps One binder 1/2 (MOB1/2)[69]. As show n in Figure 1, Lats1/2 then phosphorylates the transcriptional co-activators YAP and TAZ w hich also can function as novel sensors of the mevalonate and glycolytic pathways[70-72].
The residues Ser127and Ser397of YAP are positioned w ithin a consensus sequence(HXRXXS) phosphorylated by Lats1/2. The p hosphorylation of YAP at these sites,restricts its cellular localization to the cytoplasm, reduces its stability, and inhibits its co-transcrip tional activ ity. In ad d ition to Lats1/2, YAP and TAZ can be phosphorylated by other protein kinases[68]. Although YAP and TAZ have very similar structural top ologies, share nearly half of the overall amino acid sequence, and are thought to be largely redundant, they may differ in their regulation and downstream functions[73].
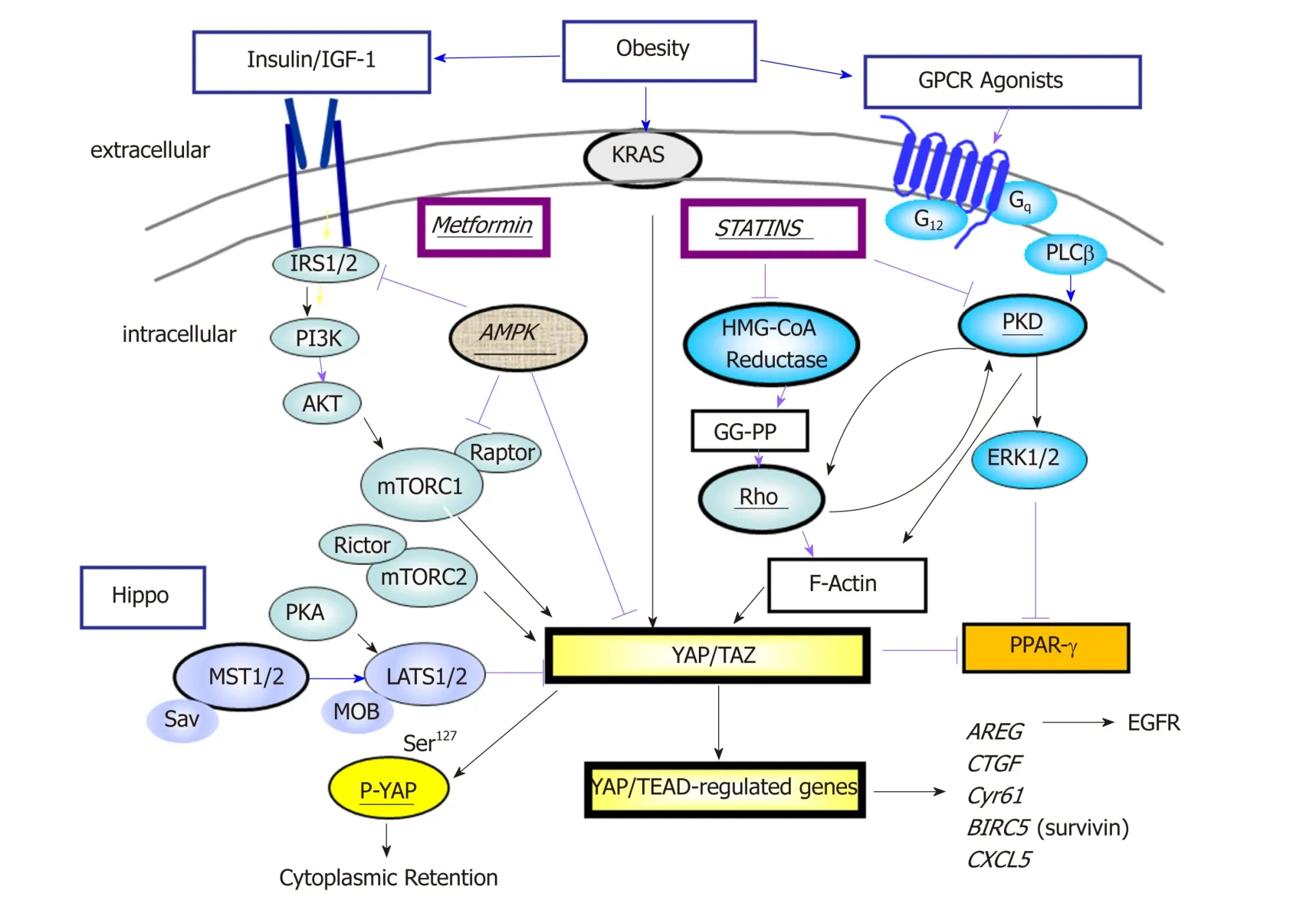
Figure 1 Yes-associated protein and WW-domain-containing transcriptional co-activator with PDZ-binding motif is a point of convergence in signaling pathways. A network that involves activated Ras, G protein-coupled receptors (GPCRs) and tyrosine kinase receptors positively regulates Yes-associated protein and WW-domain-containing transcriptional co-activator with PDZ-binding motif (YAP/TAZ) activity via Rho/PKD/organization of the actin cytoskeleton and PI3K/AKT/mTORC1. The interaction of mTORC1 and YAP is explained in the text. In addition, the localization and activity of YAP/TAZ is negatively impacted by the Hippo pathway which mediates phosphorylation of YAP and thereby its cytoplasmic sequestration. Metformin and statins inhibit YAP/TAZ activity at different sites in the network. Stimulatory effects are shown by black arrows whereas inhibitory effects are indicated by red arrows. YAP/TAZ: Yes-associated protein and WW-domaincontaining transcriptional co-activator with PDZ-binding motif; GPCR: G protein-coupled receptor.
When the Hippo pathway is not functional, YAP localizes to the nucleus where it interacts w ith the TEA-d omain DNA-bind ing transcription factors (TEAD 1-4).YAP/TEAD-regulated genes encod e for p roteins implicated many critical cellular processes, e.g., autocrine/p aracrine proliferation via EGFR (AREG) and G proteincoupled receptors (EDN 2), and interact w ith other developmental pathways activated in PDAC, including Wnt, Notch, and Hedgehog[74]. YAP/TAZ also induces epithelialto-mesenchymal transition (EMT) and induces a more und ifferentiated state to malignant cells. Accord ingly, YAP/TAZ p lay an imp ortant role in p ancreas development, w hich has implications for pancreatic regeneration, cancer, and diabetes[75]. It is accepted that YAP/TAZ acts as a potent oncogene in multiple cell types, including PDAC[76]and also contributes to the strong immunosuppressive microenvironment characteristic of mouse and human pancreatic cancer[77]. Recent findings indicate that YAP/TAZ opposes Ras-induced senescence by increasing the expression of the key enzymes involved in deoxyribonucleotide biosynthesis which are critical for DNA replication[78].
As ind icated above, YAP and TAZ do not bind directly to DNA but act by enhancing the activity of transcription factors or other proteins that interact with DNA. Although TEAD family members are the major DNA-bind ing p artners,YAP/TAZ can also bind to other transcription factors, e.g., RUNXs, p73, Smad1, Klf4,AP-1 to elicit context-specific functions[74,79,80]. It is important to point out that YAP and TAZ not only act as co-activators of transcription factors that bind to promoter sites contiguous to the gene that they control but exert regulatory effects via distant enhancer elements[81,82]. Furthermore, recent studies indicate that YAP/TAZ-bound to enhancers med iate the recruitment of the general coactivator bromodomaincontaining protein 4 (BRD4) and RNA polymerase II at promoters regulated by YAP/TAZ, thereby enhancing expression of multiple growth-regulating genes[83]. It is evident that YAP and TAZ control gene-regulatory programs through a variety of mechanism, further supporting their fundamental role in cell signaling.
Regulation of YAP/TAZ in PDAC
Recent studies d emonstrated that YAP is required for acinar-to-ductal metap lasia(ADM), an early event that precedes Pan IN p rogression into PDAC in genetically engineered mouse models[84,85]. In addition, YAP is a major mediator of pro-oncogenic mutant p53[86]and p53 deficiency p romotes YAP signaling trough Ptpn14[87]. Also,YAP confers resistance to RAF/MEK inhibitors[88]and chemotherapy in PDAC[89].While initially thought to be primarily an effector of the tumor-suppressive Hipp o pathw ay, more recent studies revealed that YAP/TAZ subcellular localization and cotranscriptional activity is regulated by multip le up stream signals includ ing those mediated by various G protein-coupled receptors (GPCRs), tyrosine kinase receptors(EGFR, MET, Insulin/IGF-1 receptor), integrins, PI3K, mTOR, PKC, PKD, RHO and actin cytoskeleton, all of w hich stimulate YAP/TAZ transcrip tional co-activator activity[66,69,76,90-92]. Recently, Src kinases, dow nstream of KRAS, have been shown to inhibit the Hip p o p athw ay by d irectly p hosp horylating Lats1 thereby activating YAP[93]. Interestingly, some of the tumor suppressive effects of w ild type p53 appear to be exerted via inhibition of YAP1 function[87].
In human PDAC cells, YAP functions as a dow nstream effector of the crosstalk betw een insulin/IGF-1 recep tor and GPCR systems[94](Figure 1). We have d emonstrated that stimulation w ith insulin and the GPCR agonist neurotensin ind uced rapid YAP nuclear import and marked ly augmented the m RNA levels of YAP/TEAD-regulated genes, including CTGF and Cyr61. The grow th-p romoting agonists regulated YAP activity via PI3K and PKD in PANC-1 and MiaPaCa-2[94],human cell lines that correspond to the squamous/quasi mesenchymal/ basal-like sub-type of PDAC. In other cell types, several studies have also been show n that PI3K activation inhibits the Hippo pathway[95,96]thereby promoting YAP activity, and PKD med iates YAP nuclear localization and activation of YAP/TEAD-regulated gene exp ression[90]. Overall, YAP has emerged as a central nod e of transcrip tional convergence in grow th-promoting signaling in PDAC cells (Figure 1). In ad dition to rap id regulation via p hosp horylation and sub-cellular localization, ad d itional pathw ays and epigenetic stimuli mod ulate YAP/TAZ protein expression. In this context, it has been show n that the RAS p athw ay, ind epend ently of the Hip p o cascad e, enhances YAP1 stability through dow nregulation of the ubiquitin ligase comp lex substrate recognition factors SOCS5/6[97]. Moreover, the eukaryotic translation initiation factor 5A (eIF5A), w hich is up-regulated by KRAS in PDAC,interacts w ith the tyrosine kinase PEAK1 leading to enhanced YAP expression[98].
The nutrient sensor mTORC1, a central dow nstream component of the PI3K/AKT and RAF/MEK/ERK pathw ays, is implicated in the development of multiple types of cancer, including PDAC[99]. Interestingly, YAP and m TORC1 form a positive feedback loop that lead s to enhanced YAP p rotein exp ression. Specifically, YAP stimulates mTORC1 via increasing the activity of the PI3K pathway[100]and augmented amino acid transport[101,102]. In turn, mTORC1 activation leads to YAP accumulation at least in part, via d ecreased autophagy[103]. Importantly, amplification and overexpression of YAP has been shown to bypass the need of mutant Kras in murine PDAC[104]and other cancer cell types[105]though the mechanism(s) differ(s), probably reflecting cell-context factors[106]. These finding indicates that YAP not only acts d ownstream of KRAS but also that YAP can sidestep the need of KRAS mutant expression in PDAC[107].
Several stud ies in d ifferent cell typ es d emonstrated that an increase in the intracellular level of cAMP inhibits YAP activation, at least in part through activation of protein kinases of the Hippo pathway[108,109]. Interestingly, concomitant expression of mutated (R201C) GNAS, w hich encodes for stimulatory G-protein alpha subunit that increases cAMP synthesis, with oncogenic Kras in mice, induced the formation of p ancreatic cystic neop lasms, resembling human intrad uctal pap illary mucinous neoplasms (IPMN), a less aggressive histological subtype of pancreatic tumors, by inhibiting YAP signaling[110]. These recent findings underscore the importance of YAP activation in the d evelop ment of sp ecifically PDAC. In this regard, it is of great interest that YAP function has been associated w ith the squamous/qu asi mesenchymal/basal-like sub-type of PDAC (d iscussed above), considered the most clinically aggressive form. The significance of YAP expression in human PDAC is discussed in the next section.
An important feature of human and murine PDAC is an extensive d esmoplastic stroma[111]that increases the stiffness of the extracellular matrix (ECM) surrounding the epithelial cancer cells[42]. The Hippo/YAP pathway has been recognized to play a critical role in mechano-transd uction[112,113]and in sensing ECM stiffness[114]but the mechanisms involved are not fully understood. Recently, the Ras-related GTPase RAP2 has been identified as a major sensor of mechanical cues from the ECM. At low stiffness, RAP2 activates the Hipp o kinases Lats1/2 thereby inhibiting YAP/TAZ activity[115]. Therefore, high stiffness lead s to inhibition of the Hip p o tumor sup p ressive p athw ay, thus enhancing the co-activator activity of YAP and TAZ.Recip rocally, increased exp ression of a number of YAP/TEAD-regulated genes,including CTGF, Cyr61 and CXCL5 contribute to shaping the stroma of PDAC, thus establishing an important amplification loop involving the tumor microenvironment leading to the stimulation of PDAC development.
YAP and human PDAC
Several stud ies reported that YAP and TAZ are over-expressed and over-active in human PDAC[104,116,117]and identified YAP expression as an ind epend ent prognostic marker for survival of PDAC[118]. We have examined the prognostic value not only of YAP but also of upstream and dow nstream components of the YAP-driven netw ork in pancreatic cancer[119]. We confirmed that higher expression of YAP is significantly associated w ith unfavorable prognosis (survival) in PDAC[120]. Ind eed, none of the patients of the population with higher levels of YAP mRNA expression survived for 5 years w hile 32% of the subset w ith the low er levels of YAP mRNA survived for 5 years or more. In ad d ition, multip le genes regulated by YAP/TEAD, includ ing AJUBA, ANLN, AREG, ARHGAP19, ARHGAP29, AURKA, BUB1, CCND1, CDK6,CXCL5, DKK1, JAG1, NOTCH2 and RHAMM w ere significantly associated w ith unfavorable prognosis in PDAC[120]. In a further analysis of the data, we verified that the expression of each of these genes was positively and significantly correlated w ith the expression of YAP in PDAC. In contrast, genes in pathways, e.g., LKB/AMPK and cAMP/PKA, that oppose YAP action, including STRAD, MARK1, PKA, are associated w ith favorable prognosis in PDAC p atients[120]. Similar results w ere obtained using other w eb-based tools, such as Gene Expression Profiling Interactive Analysis[121].
YAP and obesity
Besid es its recognized role in the regulation of grow th and d evelopment, recent studies show that Hip po kinases and YAP/TAZ transcrip tional coactivators, are regulated by metabolism and conversely that the Hipp o/YAP pathw ay controls metabolic processes in physiological and pathologic conditions, including obesity and T2DM[122,123]. In fact, cellular metabolites and metabolic pathways, e.g., glucose and free fatty acid s, regulate the Hippo pathw ay. Glucose metabolism through the glycolytic pathw ay activates phosp hofructokinase 1 (PFK1), a key rate-limiting enzyme of glycolysis. In turn, PFK1 interacts w ith TEAD, thereby regulating YAP/TEAD complex formation and expression of YAP/TEAD-regulated genes[70]. Furthermore, Olinked β-N-acetylglucosamine (O-GlcNAc) is another post-translational mechanism by w hich a sugar is attached to serine residues of nuclear or cytoplasmic proteins and mod ifies protein activity[124]. Ind eed, the attachment of O-GlcNAc to Ser109of YAP stimulates its transcriptional co-activator activity by interfering w ith the interaction of YAP w ith Lats1/2, thus p rotecting YAP from inhibitory p hosp horylation and provid ing a novel mechanism linking glucose availability to YAP activity[125]. This multilayered regulation of YAP activity by glucose metabolism is p otentially important in the obese state, which often is accompanied by insulin resistance and elevated glucose levels.
A characteristic and defining feature of obesity is the enlargement of adipose tissue d ep ots, w hich is often accomp anied by ad ip ose tissue (AT) inflammation[126].Dysfunctional ad ip ose tissue w ith alterations of adip okine production, ectop ic fat storage, and AT inflammation are thought to be critical, pathophysiological processes und erlying the d evelop ment of insulin resistance. Ad ip ocytes and ad ipose tissue macrophages are central cellular p layers of AT inflammation[127-132]. The Hip p o pathw ay has been show n to modulate adip ocyte proliferation and d ifferentiation,w ith YAP/TAZ nuclear localization stimulating p roliferation and sup p ressing ad ipogenesis[133-136]. As d epicted in Figure 1, nuclear YAP/TAZ interacts w ith and inhibits PPAR-γ, a major pro-ad ipogenic transcription factor, thereby suppressing adipocyte differentiation[133,137]. In that context, hyperglycemia and advanced glycation end products impair adipogenesis by upregulating and activating YAP[138].
There are few studies investigating the importance of YAP/TAZ in macrophage polarization[139]. It has been show n that the cell shap e, ind ep end ent of cytokines present in the micromilieu, has a profound influence on macrophage polarization via the actin cytoskeleton[140], w hich strongly suggests an important role of YAP/TAZ in this p rocess d ue to the critical function or YAP/TAZ as mechano-sensors and mechano-transd ucers[112,113]. In ad d ition, ad ip ose tissue in obese subjects is characterized by p eri-ad ip ocyte fibrosis w ith elevated levels of CTGF (connective tissue growth factor)[141], a recognized product of YAP/TEAD transcriptional activity.Our ow n studies have show n that YAP is overexpressed in mesenteric adipose tissue of obese KC mice (unpublished). Taken together, an important role of YAP/TAZ in ad ip ose tissue inflammation d uring obesity emerges, w hich might have important implications in the PDAC promoting effects of obesity[142,143].
Strategies to inhibit YAP/TAZ in pancreatic cancer
As indicated above, YAP hyper-activation can evade the need of KRAS mutant expression in PDAC[107]. Thus, even if Ras could be effectively inhibited by new therapies, YAP amplification could provide a potential pathway to tumor recurrence.Given that YAP is as a key element not only d ow nstream of Ras but also an alternative route to bypass the need of this oncogene for tumor relapse, YAP is emerging as a fundamental target in PDAC. Although targeting transcription factors or their co-activators has proven difficult, recent studies suggest novel approaches to inhibit YAP/TAZ activity with drugs in clinical use, including statins and metformin in PDAC and other malignancies.
Statins
Several stud ies d emonstrated activation of the p athw ay lead ing to mevalonate biosynthesis in epithelial cancers through mutant p 53[144-146]and AKT/m TORC1[146].Statins, w hich have been used to treat d yslipid emia and prevent heart d iseases,selectively inhibit 3-hyd roxy-methylglutaryl (HMG) CoA red uctase[147], the ratelimiting enzyme in the generation of mevalonate (Figure 2). Mevalonate is a precursor for the generation of important lip ids and lipid intermediates, including farnesyl pyrophosp hate (FPP), geranylgeranyl pyrophosphate (GG-PP) and cholesterol. The function and activity of small GTPases of the Rho family, including Rho A and C,depend on the transfer of the geranylgeranyl moiety of GG-PP to a cysteine in their COOH-terminal region. Active Rho p lays a critical role in YAP/TAZ activation through actin remod eling in several cell p op ulations[69](Figure 2). Accord ingly,increased expression of RHOA and RHOC is associated with unfavorable prognosis in patients with PDAC.
Numerous epid emiological stud ies have concluded that statin use is correlated w ith beneficial effects in PDAC[148-156], esp ecially in men[151,152]. A large stud y demonstrated that statins w ere associated with a significantly reduced PDAC risk (by 34%) with a stronger effect in males[151]. The beneficial effects of statins depend on the type of statins used, w ith several reports show ing positive associations with lipophilic(and not hydrophilic) statins and reduced cancer risk[157-160]. However, a recent study,in w hich statin use w as self-reported and the type of statins w as not documented in early cohorts, failed to detect an effect of statins in low ering PDAC risk[161]. The same authors published a follow up study of the same dataset, in which they reported an increased survival in PDAC patients w ith regular pre-d iagnosis use of statins[162].Recently, a meta-analysis of PDAC risk that included more than 3 million participants and 170000 pancreatic cancer patients has been published[163]. This study indicates a significant d ecrease in p ancreatic cancer risk w ith statin use, thus reinforcing the conclusion that statin administration is associated w ith beneficial effects in PDAC patients. In addition to their potential efficacy in primary prevention and interception,statins may improve the outcome of patients after surgical removal of their primary PDAC[148,149,164], ind icating a p ossible role of statins in the p revention of PDAC recurrence.
In preclinical studies[165,166], statins d elayed progression of PDAC in mice harboring KrasG12D. Statins w ere identified as potential YAP inhibitors by screens of molecules that changed the nuclear/cytoplasmic distribution of YAP[167]. Our ow n experiments using PDAC cells also indicated that lipophilic statins induce cytoplasmic localization of YAP and marked ly inhibited YAP/TEAD-regulated genes, p roliferation, and colony formation by PDAC cells (submitted for p ublication). Taken together,converging evid ence from ep id emiological and p reclinical stud ies ind icates a protective effect of statins in PDAC.
Metformin
1,1-dimethylbiguanide hydrochloride (metformin) is the most widely administered drug for the treatment of T2DM worldwide[168,169]. The anti-diabetic, i.e., lowering the blood glucose levels, actions of metformin are mediated systemically by a reduction of hepatic glucose production and output into the circulation and improvement of insulin sensitivity via increasing cellular uptake of glucose in skeletal muscles and adipose tissue[170]. In addition to lowering blood glucose levels, metformin decreased the levels of insulin and IGF-1 in both diabetic and non-diabetic patients[171,172].Multiple epidemiological studies showed an association of metformin with reduced incidence, recurrence and mortality of cancer in patients with T2DM[173-182]. However, a therapeutic efficacy of metformin has not been observed in all studies[183], in particular in late-stage, advanced cases of cancer. In that context, recent meta-analyses supported the notion that the beneficial effects of metformin depend on the stage of the tumor, w ith a substantially enhanced survival in patients with local, nonmetastatic, disease[178,184]. Further reports indicated that metformin administered to T2DM patients could be also beneficial in secondary chemoprevention, i.e. after surgical resection of the cancer in the pancreas[185,186].
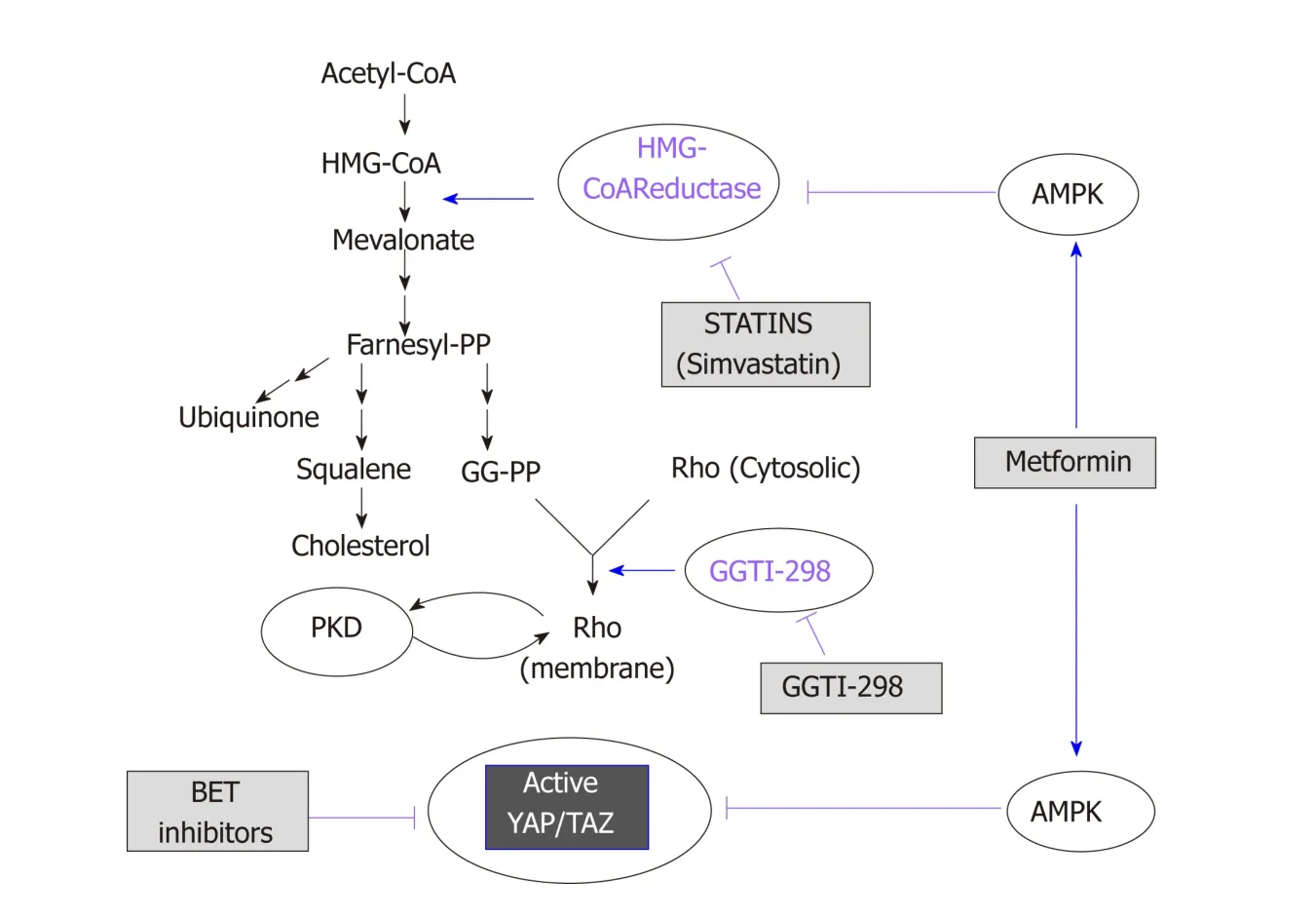
Figure 2 Schematic overview of the mevalonate pathway. The scheme illustrates the site of action of statins,metformin and bromodomain and extra-terminal domain inhibitors (see text for details). BET: Bromodomain and extra-terminal domain; AMPK: AMP-activated protein kinase; GG-PP: Geranylgeranyl pyrophosphate.
The mechanism of action of metformin remains incompletely understood. Besides its systemic (glucose low ering) effects, at the cellular level metformin indirectly activates AMP-activated protein kinase (AMPK)[187], though other AMPK-independent mechanisms are also operational[188,189]. AMPK is activated by phosphorylation by the tumor suppressor LKB-1/STK11 in the activation loop[190]when cellular ATP levels decrease and 5'-AMP and ADP concentrations increase[169]. It generally thought that metformin lead s to AMPK activation by directly inhibiting complex I of the mitochondrial respiratory chain[191,192], which leads to a decrease in ATP synthesis resulting in increased AMP and ADP thereby leading to AMPK activation. AMPK supp resses cellular p roliferation by inhibiting the function of m TORC1 through several mechanisms. AMPK activates TSC2 by phosphorylation on Ser1345[193-195], which leads to an accumulation of inactive Rheb-GDP thereby inhibiting m TORC1. AMPK can also inhibit m TORC1 function by phosphorylation of Raptor, w hich disrupts its complex w ith m TOR[196]. In ad dition, m TORC1 activation ind uced by insulin/IGF-1 signaling is also inhibited via p hosp horylation of IRS-1 on Ser794by AMPK, a phosphorylation that impedes PI3K activation[197,198]. We demonstrated that metformin,at low concentrations, activates AMPK in PDAC cells[199,200]and inhibits mTORC1, ERK and DNA synthesis via AMPK[199-201]. Metformin also reduced the rate of grow th of PDAC xenografts[202,203]. Furthermore, we recently reported that oral administration of metformin strikingly prevented the increase in PDAC incidence in KC mice with dietind uced obesity[204]. This effect w as associated with an increase in pancreatic AMPK activity (as measured by ACC Ser79p hosp horylation), and d ecrease in p hosp ho MEK1/2 (Ser217/221), phospho S6 (Ser235/236), and phospho ERK1/2 (Thr202, Tyr204)[204]. In that context, berberine, a natural compound that activates AMPK and inhibits ATP p rod uction, also inhibited m TORC1, ERK, DNA synthesis and p roliferation of pancreatic cancer cells and reduced the growth of PDAC xenografts[201].
Recent evid ence ind icates that AMPK also opp oses YAP activity via multip le mechanisms, includ ing d irect YAP p hosphorylation on Ser94[205,206], a residue that is important for the interaction of YAP with TEAD. In addition, AMPK has been shown to phosp horylate HMG-CoA red uctase (Ser872), thereby inhibiting its activity and red ucing mevalonic acid synthesis[207]. Furthermore, AMPK p hosp horylates and activates upstream regulators of the Hipp o p athw ay[208]. The inhibitory effects of AMPK on the YAP/TAZ pathw ay is illustrated in Figure 2. These studies suggest an imp ortant d irect link among ad enine nucleotid e levels, AMPK and YAP/TAZ activity. In studies from our laboratories, w e found recently that diet-induced obesity markedly increased pancreatic TAZ expression in KC mice and that oral metformin prevented the increase in YAP/TAZ[204]. Given that statins and metformin inhibit YAP activation through different mechanisms, it is logical to speculate that administration of a combination of these FDA-approved d rugs will suppress YAP/TAZ activity and exerts PDAC-protective activity. The scheme presented in Figure 1 dramatizes this notion by showing that statins and metformin reach YAP through different pathways.
Inhibitors of BRD4: A new approach for targeting YAP/TAZ
BRD4, w hich interacts with acetyl-lysine, acts as a critical regulator of the expression of selected subsets of genes. Bromod omain and extra-terminal d omain (BET)inhibitors interfere with the proliferation of PDAC cells, raising the possibility that BET p roteins may be new targets for PDAC therap y[209]. A recent elegant stud y d emonstrated a d irect p hysical interaction betw een YAP or TAZ and BRD4, as revealed by co-immunoprecip itation experiments. The d ata imp ly that YAP, TEAD and BRET-containing proteins (e.g., BRD4, BRD2) form a multi-molecular complex in the nucleus[83]. Consistent with the notion that BRD4 plays a critical role in YAP/TAZ function, the BET cell-permeable inhibitor JQ1[210]dow nregulates the expression of YAP/TAZ-regulated genes[83]. Consid erable efforts are being mad e to d evelop new inhibitors of BRD proteins and thus this field w ill d evelop rap id ly[211]. These new find ings suggest a novel ap proach to target YAP/TAZ that remains to be tested exp erimentally in vivo, using mod els of PDAC. As suggested by Figure 2, the possibility of using BET inhibitors in combination w ith statins and/or metformin is attractive and warrants further experimental work.
Feedback loops and effect of pathway inhibitors
Most signaling p athw ays are subjected to p otent feed back loop s that ad just the activity and function of the signaling netw ork. There is evid ence that besides their stimulating effects on mitogenic signaling the m TORC1/S6K and RAF/MEK/ERK pathw ays also med iate robust negative feed back loop s that restrict the activity of insulin/IGF-1, EGFR, and other tyrosine kinase receptors[99]. In that context, the m TORC1/S6K pathw ay inhibits the function of IRS-1 by phosphorylating several residues (Ser636/639by mTORC1 and Ser307/636/1001by S6K)[212]. Inhibitors of mTORC1/S6K or MEK/ERK suppress these feedback loops, w hich in turn causes a compensatory activation of upstream signaling molecules, e.g., PI3K, AKT, and ERK that as a consequ ence strongly counteract the anti-p roliferativ e actions of these inhibitors[99,200,213]. The up-regulation of these pathw ays conceivably can promote YAP activity leading to drug resistance. Therefore, a detailed understanding of feed back mechanisms that regulate up stream signaling is critical and w ill enable the identification of rational drug combinations that w ill circumvent d rug resistance produced by unleashing the activity of alternative pathw ays.
CONCLUSION
Despite major ad vances in d efining the molecular mutations d riving PDAC, this disease remains universally lethal w ith an overall 5-year survival rate of only about 7%-8%. More efficacious therapeutic strategies are clearly needed but given the late presentation and early d issemination of the d isease, substantial efforts should be concentrated on prevention and interception. Hereby, d etailed know led ge of the molecular mechanisms und erlying risk-factor promoted PDAC w ill surely facilitate and enable the d iscovery of novel molecular targets and agents for p rimary or secondary prevention. Epidemiological studies convincingly demonstrate that obesity is a risk factor for PDAC development, the importance of which takes an added level given the epidemic proportions of metabolic diseases. It is also recognized that almost all PDACs harbor an oncogenic KRAS mutation, w hich seems necessary but not sufficient for complete PDAC formation. Besides additional mutations, which greatly accelerate PDAC progression in mice, environmental conditions, includ ing obesity,T2DM, and inflammation, have been shown to also promote PDAC in murine models.As illustrated in Figure 1, w e propose that PI3K/mTORC1 and PKD/ERK are critical nod es in the netw ork activated by GPCRs, EGFR and insulin/IGF-1 receptor in PDAC. These signaling modules are responsive to obesogenic signals and reinforce KRAS signaling. In turn, oncogenic KRAS mutations p otentiate the intensity of signaling netw ork emanating from GPCRs, EGFR, and insulin/IGF-1 receptors by activating PI3K/AKT and Raf/MEK/ERK, the most p rominent d ow nstream pathways of oncogenic KRAS.
We also postulate that YAP/TAZ transcriptional co-activators are central and critical players in this amplification netw ork, further intensifying positive feed back loops. GPCRs, EGFR, and insulin/IGF-1 receptor signaling rapidly stimulate nuclear import and transcriptional co-activator activity of YAP/TAZ, while oncogenic KRAS increases the levels of YAP p rotein. In turn, YAP stimulates signaling via autocrine/paracrine stimulation of EGFR via increased production of EGFR ligands(e.g., amphiregulin), thereby further propagating and enhancing KRAS activity, as w ell as creating an immunosup pressive microenvironment. We hyp othesize that oncogenic KRAS potentiates a signaling netw ork that is stimulated and sustained by environmental factors. As YAP/TAZ play a central role in the signaling netw ork,targeting this network at d ifferent sites with FDA-approved drugs, including statins and metformin (Figure 2), is therefore a comp elling app roach, esp ecially in obese patients at higher risk of developing PDAC.
杂志排行

World Journal of Gastroenterology的其它文章
- Repurposing drugs to target nonalcoholic steatohepatitis
- Considerations of elderly factors to manage the complication of liver cirrhosis in elderly patients
- Lysyl oxidase and hypoxia-inducible factor 1α: biomarkers of gastric cancer
- Predictive and prognostic implications of 4E-BP1, Beclin-1, and LC3 for cetuximab treatment combined with chemotherapy in advanced colorectal cancer with wild-type KRAS: Analysis from real-world data
- Extract of Cycas revoluta Thunb. enhances the inhibitory effect of 5-f luorouracil on gastric cancer cells through the AKT-mTOR pathway
- Unconjugated bilirubin alleviates experimental ulcerative colitis by regulating intestinal barrier function and immune inflammation