加米霉素对照品标定方法的建立
2019-04-09张聪姚路路侯林樊丽博杨会鲜周德刚张晓会
张聪,姚路路,侯林,樊丽博,杨会鲜,周德刚,张晓会
(1.国家兽用药品工程技术研究中心,河南洛阳 471000;2.洛阳惠中兽药有限公司,河南洛阳 471000)
加米霉素是法国梅里亚公司开发研制的新一代半合成十五元氮杂环大环内酯类抗菌药物,其注射液于2017年通过农业部进口兽药注册在我国上市销售,用于治疗和预防由溶血性曼氏杆菌、多杀性巴氏杆菌、睡眠嗜组织菌等主要病原菌引起的牛呼吸系统疾病[1]。目前国内有多家单位开展加米霉素及注射液的仿制研究[2-4],但加米霉素法定对照品在国际和国内并无单位提供,困扰了加米霉素原料及其注射液的质量控制,本文研究建立了加米霉素对照品的标定方法,为其对照品的标定提供技术依据。
1 仪器与试药
1.1 仪器与试剂 高效液相色谱系统(Waters e2695型HPLC;2998型PDA检测器;Empower 3色谱工作站软件),美国Waters公司;XBridge C18柱(250 mm×4.6 mm,5 μm),美国Waters公司;AB265-S电子分析天平,梅特勒托利多公司;Seven easy型酸度计,梅特勒托利多公司;GZX-9070MBE型电热鼓风干燥箱,SX2-2.5-10型箱式电阻炉,上海博迅实业有限公司医疗设备厂;磷酸氢二钠、磷酸为分析纯;甲醇、乙腈为色谱纯;水为超纯水。
1.2 试药 加米霉素对照品原料,由洛阳惠中兽药有限公司制备。
2 方法与结果
2.1 结构确证
2.1.1 采用紫外光谱(UV)、红外光谱(IR)、质谱(MS)、核磁共振波谱(NMR)对加米霉素原料进行结构确认 测试方法及条件:UV测定,供试品用乙醇溶解,浓度为0.011 mg/mL,扫描范围190~400 nm;IR测定,采用KBr压片法,供试品与KBr的比例约为1∶200;1H-NMR测定为500 MHz,13C-NMR测定为500 MHz,供试品采用CDCl3溶解;MS测定,离子化方式为ESI+。
结构确认结果如下:UV λmax:204 nm;IR v:3534、2969、2937、2876、2831、2795、1735、1637、1458、1421、1380、1349、1295、1279、1249、1166、1135、1109、1094、1075、1058、1016、1004、983 cm-1;ESI-MSm/z:777.54 [M+H]+;1H NMR(CDCl3):δ 0.89, 0.98, 0.99, 1.08, 1.22, 1.30, 1.54, 1.70, 1.91, 2.01, 2.30, 2.74, 3.01, 3.14, 3.29 3.55, 3.62, 4.08, 4.23, 4.38, 5.00。13C NMR(CDCl3):δ177.60, 105.09, 96.47, 77.92, 77.30, 7.05, 76.79, 75.31, 74.27, 72.79, 70.39, 69.23, 65.60, 64.97, 59.27, 49.37, 46.87, 42.48, 40.37, 34.97, 32.53, 28.95, 22.06, 21.55, 21.16, 17.82, 16.02, 15.23, 14.54, 12.92, 12.09, 11.35。以上数据与文献[3,5]报道数据一致,表明对照品原料化学结构与加米霉素相同。
2.1.2 采用X射线单晶衍射法对加米霉素原料进行结构确认 单晶培养条件:取对照品原料0.1 g,置洁净容器内,加乙腈溶解,室温放置,缓慢挥发溶剂至晶体长成。
对照品原料X射线单晶衍射测定结构单元见图1,绝对构型与加米霉素相同。
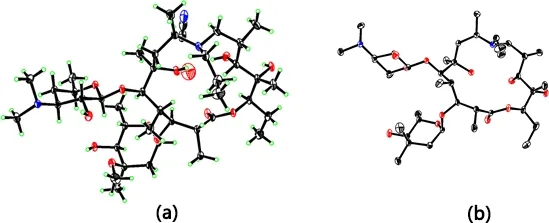
a含溶剂的结构单元 b去除氢的结构单元a Structural Unit Containing Solvents b Structural unit for hydrogen removal图1 对照品原料结构单元 Fig 1 Structure Unit of reference material
2.2 HPLC纯度检查
2.2.1 色谱条件 参考欧洲药典EP8.0版阿奇霉素有关物质检查方法[6]并进行优化,采用Waters XBridge C18(250 mm×4.6 mm 5 μm)色谱柱;流动相A为0.01 mol/L磷酸氢二钠溶液(稀磷酸调pH值至8.9)-甲醇-乙腈(90∶2.5∶7.5),流动相B为0.01 mol/L磷酸氢二钠溶液(稀磷酸调pH值至8.9)-甲醇-乙腈(28∶18∶54),线性梯度洗脱,柱温30 ℃;流速为1.0 mL/min,检测波长为205 nm。
2.2.2 供试品测定 称取对照品原料适量,加稀释液[1.73 g/L磷酸二氢铵溶液(氨水调pH值至10.0)-甲醇-乙腈(35∶35∶30)]溶解并稀释制成每1 mL中约含10 mg的溶液,作为供试品溶液。精密量取供试品溶液20 μL,注入液相色谱仪,记录色谱图。扣除保留时间4 min前的溶剂色谱峰,按峰面积归一化法计算纯度,结果见图2和表1。
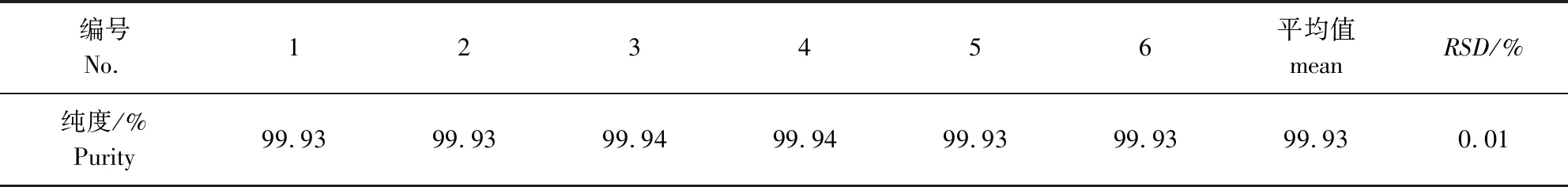
表1 纯度测定结果Tab 1 The results of purity test

图2 对照品原料纯度测定色谱图Fig 2 The chromatogram of reference material Purity test
2.3 干燥失重 按《中国兽药典》2015年版一部[7]干燥失重测定法(附录0831)测定,结果见表2。
2.4 炽灼残渣 按《中国兽药典》2015年版一部[7]炽灼残渣测定法(附录0841)测定,结果见表3。

表2 干燥失重测定结果Tab 2 The results of Loss on drying

表3 炽灼残渣测定结果Table 3 The results of residue on ignition
2.5 定量赋值 加米霉素对照品原料定值采用国际通用的质量平衡法确定含量,同时采用定量NMR技术对质量平衡法赋值准确性进行验证。
2.5.1 质量平衡法赋值结果 含量(%)=纯度×(1-干燥失重-炽灼残渣)×100%,结果为99.93%×(1-1.05%-0.10%)×100%=98.8%。
2.5.2 定量NMR法验证结果 依据《中国药典》2015年版四部[8]通则0441核磁共振波谱法,试验采用内标法,以马来酸作为内标,样品用氘代乙醇溶解后,以加米霉素1H谱中的2.66(3H)信号峰与内标物1H谱中的6.26(1H)信号峰(图3)计算供试品的含量,定量NMR法含量测定结果为98.3%。
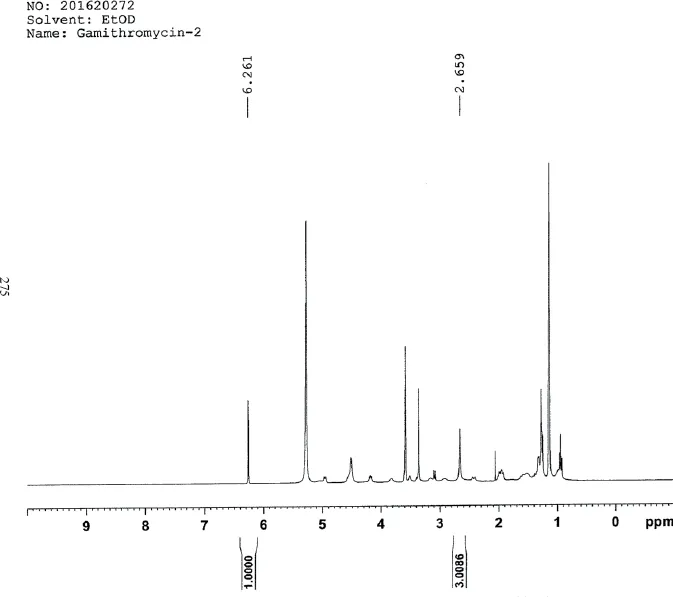
图3 加米霉素定量核磁共振光谱图Fig 3 The spectrogram of Quantitative nuclear magnetic resonance
2.6 对照品原料稳定性考察 加米霉素对照品原料,用安瓿瓶分装,50 mg/瓶,放入药品稳定性试验箱中,在(40±2)℃、相对湿度75%±5%条件下放置6个月,分别于第1、2、3、6月取样,对样品性状、熔点、纯度进行检查,结果见表4。
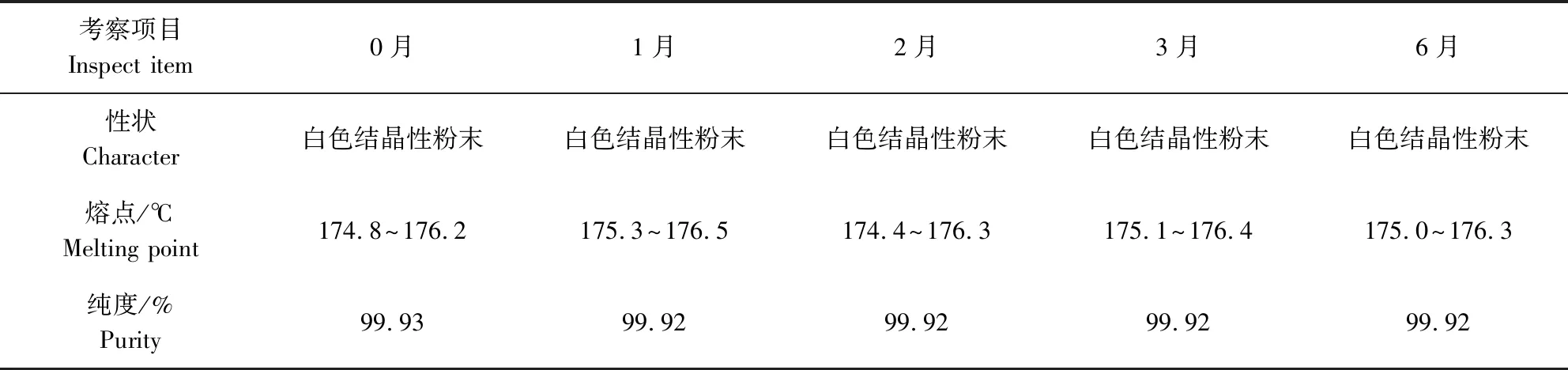
表4 稳定性考察结果Tab 4 The results of stability investigation
3 讨论与结论
3.1 加米霉素结构确证 加米霉素为十五元氮杂环大环内酯类化合物,化学结构中具有较多的手型碳原子,因此,单纯采用核磁共振方法难以确定每个手型中心的绝对构型,本研究通过溶剂结晶方法培养得到加米霉素单晶,采用X-射线单晶衍射方法对加米霉素的绝对构型进行了测定。
3.2 质量平衡法赋值 本研究按质量平衡法对本批加米霉素对照品原料进行了含量定值[9]。根据质量平衡法原理,标准品含量(%)=[100%-水分(%)-残留溶剂(%)-无机杂质(%)]×纯度(%),当用干燥失重方法可将标准品中的水分和残留溶剂全部挥发出来并使供试品达到恒重时,本公式中的水分和残留溶剂的测定结果可用干燥失重结果代替[10]。加米霉素对照品原料最后采用丙酮-水系统进行精制,在105 ℃能全部挥发出去,试验证明加米霉素对照品原料在该温度能很快干燥至恒重,说明水分和有机溶剂能完全挥发,可用干燥失重结果代替水分和残留溶剂的测定。
3.3 含量定值 为了准确标定加米霉素对照品原料的含量,采用了两种不同原理的方法进行测定。由于原研公司加米霉素质量标准并未公开,根据其结构特征,参考泰拉霉素质量标准[11]及各国家阿奇霉素药典标准[6,12],本研究建立了加米霉素HPLC纯度检测方法,并对采用的方法进行了方法学验证,以确保纯度检测结果的准确性。结果表明质量平衡法结果与定量NMR法结果相对偏差为0.3%,两种方法标定结果一致。
本批加米霉素对照品原料的结构鉴定结果与其结构相符合,纯度大于99%(HPLC法),证明该批原料可以用于加米霉素对照品的制备。采用质量平衡法测得的加米霉素对照品原料含量为98.8%,用定量NMR法测得的结果为98.3%,两种方法测定结果差异较小,说明本批加米霉素对照品原料赋值结果准确,可作为加米霉素原料及其制剂研制过程中对照品使用。
