HPLC法测定不同外观形态的桂枝中肉桂酸的含量
2019-04-03杨羽君鄂秀辉
杨羽君,鄂秀辉
(天津市天士力控股集团有限公司研究院,天津 300410)
桂枝为樟科植物肉桂(CinnamomumcassiaPresl)的干燥嫩枝,具有发汗解肌、温通经脉、助阳化气、平冲降气之功效[1]。自汉代张仲景《伤寒论》以来,为历代医家所常用。据统计,桂枝在经方中的使用频率仅次于甘草,是常用的解表药,主要产地为广东、广西、云南等省区[2]。临床上多用于风寒感冒、脘腹冷痛、血寒经闭、关节痹痛、痰饮、水肿、心悸、奔豚等病症[3]。
桂枝中含有的主要成分为桂皮醛(Cin-namaldehyde)和肉桂酸(Cinnamic acid)。药理研究表明:肉桂酸具有抗菌、消炎作用[4]。《中国药典》2015年版(一部) 桂枝含量测定项下仅对桂皮醛做了相关规定,未对肉桂酸做出规定。又由于目前药材市场的桂枝商品外观形态不同,为更加客观、全面地评价桂枝质量,本文建立了测定桂枝中肉桂酸含量的方法,并分析市售多批桂枝,发现其外观形态与成分含量、价格之间的关系,为不同形态的桂枝饮片提供质量控制的依据。
1 仪器与试药
1.1仪器 高效液相色谱(Waters HPLC e2695-2998,PDA 检测器);电子分析天平(XS205,瑞士METTLER TOLEDO 公司);离心机(ST16R,美国Thermo公司);超纯水系统(Millipore-Q,Millipore公司);超声波清洗器(KQ-500DV,昆山市超声仪器有限公司)。
1.2试药 肉桂酸对照品(批号110786-200503,中国食品药品检定研究院)。甲醇(色谱纯,德国Merck公司);乙腈(色谱纯,德国Merck公司);超纯水(Millipore-Q制备);冰乙酸(天津市致远化学试剂有限公司)。12批不同产地的桂枝饮片(批号S1~S12,购于安国药材市场),由天士力研究院现代中药开发中心人员鉴定为樟科植物肉桂(CinnamomumcassiaPresl)的干燥嫩枝炮制而成的饮片。
2 方法与结果
2.1色谱条件 采用Agilent ZORBAX Eclipse C18色谱柱(250 mm×4.6 mm,5 μm),以乙腈(A)-0.1%乙酸水(B)为流动相,梯度洗脱,0~12 min,40%~43% A;12~20 min,43% A;检测波长:276 nm;流速:1.0 ml/min;柱温:30 ℃;进样量:10 μl;理论板数按肉桂酸峰计算应不低于3 000。
2.2溶液制备
2.2.1对照品溶液的制备 取肉桂酸对照品适量,精密称定,加甲醇制成每1 ml含10 μg的溶液,即得。
2.2.2供试品溶液的制备 取桂枝粉末(过四号筛)约200.0 mg,精密称定,置具塞锥形瓶中,精密加入甲醇25.0 ml,称定重量,超声处理(功率250 W,频率40 kHz)30 min,放冷,再称定重量,用甲醇补足减失的重量,4 900 r/min离心10 min,精密量取上清液1.0 ml,置5 ml量瓶中,即得。
2.3系统适用性试验 取对照品溶液和供试品溶液务10 μl,按“2.1”项下色谱条件进样测定,结果肉桂酸色谱峰峰形较好,且与杂质峰较好分离。见图1。
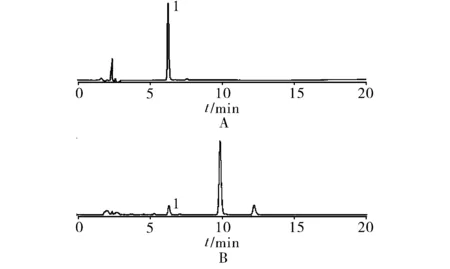
1.肉桂酸
2.4线性关系的考查 取肉桂酸对照品5.0 至50 ml量瓶中,精密称定,加甲醇定容至刻度,摇匀,滤过,精密吸取续滤液10.0 ml置100 ml量瓶中,加甲醇定容至刻度,摇匀,滤过,即得对照品溶液A。精密吸取对照品溶液A 3.0 ml于5 ml量瓶中,加甲醇定容至刻度,摇匀,滤过,即得对照品溶液A1;精密吸取对照品溶液A 2.0 ml于5 ml量瓶中,加甲醇定容至刻度,摇匀,滤过,即得对照品溶液A2;精密吸取对照品溶液A 3.0 ml于10 ml量瓶中,加甲醇定容至刻度,摇匀,滤过,即得对照品溶液A3;精密吸取对照品溶液1.0 ml于5 ml量瓶中,加甲醇定容至刻度,摇匀,滤过,即得对照品溶液A4;精密吸取对照品溶液A 1.0 ml于10 ml量瓶中,加甲醇定容至刻度,摇匀,滤过,即得对照品溶液A5;精密吸取对照品溶液A 1.0 ml于25 ml量瓶中,加甲醇定容至刻度,摇匀,滤过,即得对照品溶液A6。按照“2.1”项下色谱条件,分别精密吸取对照品A~A6溶液,注入液相色谱仪,记录色谱峰面积,以峰面积为纵坐标(Y),质量浓度(μg/ ml)为横坐标(X)绘制标准曲线,得回归方程:Y= 85 642X+4 440.8(r=0.999 8),结果表明,肉桂酸在0.40~9.98 μg/ml范围内线性关系良好。
2.5精密度试验 取对照品溶液,按照“2.1”项下的色谱条件测定,连续进样6次,记录峰面积,计算肉桂酸峰面积的RSD值为0.39%。结果表明,方法的日内精密度好。
2.6重复性试验 精密称取同一批桂枝粉末(过四号筛)200.0 mg 6份。按照“2.2.2”项下的方法制备供试品溶液,按照“2.1”项下的色谱条件进行测定,记录峰面积,计算肉桂酸含量的RSD值为0.79%。结果表明,方法重复性好。
2.7稳定性试验 取供试品溶液,分别于配制后第0、1、4、15、18、24和48 h进样,按照“2.1”项下的色谱条件进行测定,记录峰面积,计算肉桂酸含量的RSD值为0.96%。结果表明,供试品溶液在48 h内稳定性好。
2.8加样回收率试验 平行称取同一批桂枝粉末(过四号筛)100.0 mg 9份,精密称定,分为高、中、低三个水平,每个水平各3份,分别加入一定量对照品,按照“2.2.2”项下的方法制备供试品溶液,按照“2.1”项下的色谱条件进行测定,记录峰面积,计算回收率,结果平均回收率为98.18%,RSD为1.13%。见表1。
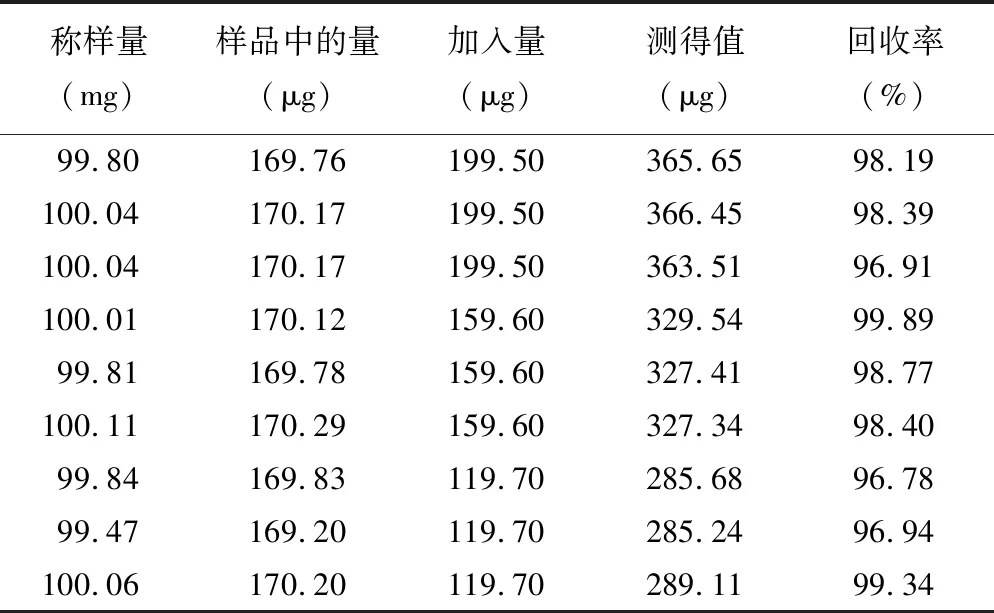
表1 肉桂酸加样回收率试验结果
2.9样品含量测定 取12批桂枝粉末(过四号筛)每批各2份,每份200.0 mg,精密称定,按照“2.2.2”项下方法制备供试品溶液,按照“2.1”项下的色谱条件进行测定,记录色谱图和峰面积,运用外标法计算含量,结果见表2。本试验测定桂枝12批,肉桂酸含量分布在0.112 2%~0.232 3% 之间。

表2 12批桂枝中肉桂酸含量测定结果(n=2)
3 讨论
3.1色谱条件的考查
3.1.1检测波长的确定 取肉桂酸对照品及供试品分别进行210~400 nm全波长紫外扫描,采用二极管阵列检测器(PDA),获得在不同波长下的数据。检测到肉桂酸最大吸收波长为276 nm,故选择276 nm作为肉桂酸的检测波长。
3.1.2流动相的确定 本试验先后考查了乙腈-水、乙腈-0.1%磷酸水、乙腈-0.1%乙酸水和乙腈-1%乙酸水体系,结果显示肉桂酸在加酸条件下出峰效果更好。乙腈-水体系基线稳定,未将色谱峰分离,理论塔板数过低;乙腈-0.1%磷酸水体系基线稳定,系统压力较高;乙腈-1%乙酸水体系基线稳定,系统压力小,但流动相pH过低,长期使用影响色谱柱及仪器;乙腈-0.1%乙酸水体系基线稳定,系统压力小,峰型好。故选择乙腈-0.1%乙酸水作为流动相,进行梯度洗脱。
3.1.3柱温的确定 本试验分别在柱温25、30和35 ℃进样,考查色谱参数优势。结果表明,柱温为30 ℃时对称因子、理论板数和分离度最优,所以柱温选择30 ℃,结果见表3。

表3 肉桂酸含量测定中不同柱温考查结果
3.2提取条件的考查
3.2.1提取时间的确定 取同一批桂枝粉末(过四号筛)200.0 mg 4份,精密称定,分别置具塞锥形瓶中。分别精密加入甲醇50.0 ml,称定重量。分别超声处理(功率250 W,频率40 kHz)20、30、40和50 min,放冷,再按照“2.2.2”项下方法配制,即得4份供试品溶液。按照“2.1”项下的色谱条件测定。发现超声20 min含量明显偏低,说明提取未完全;超声40和50 min时含量较超声30 min含量仅有微量的波动,但在1.5%左右,故判断为系统误差。本着操作简便、节约等原则确定提取时间为30 min。结果见表4。
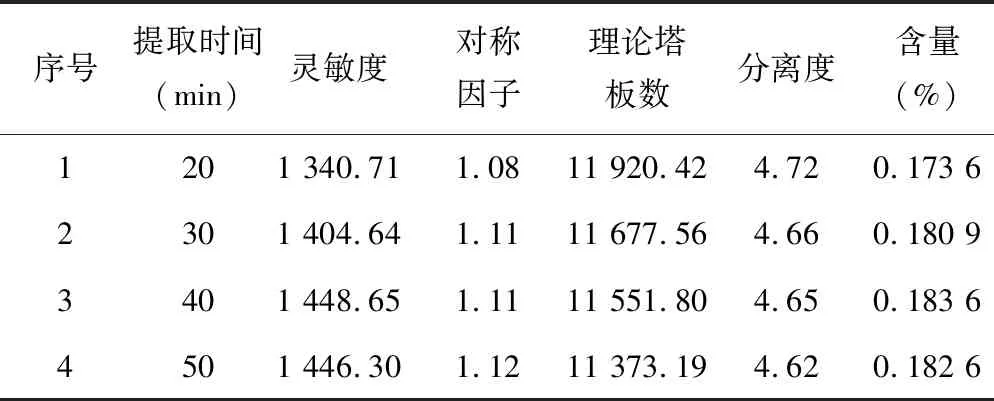
表4 不同提取时间考查结果
3.2.2提取溶剂体积的确定 取同一批桂枝粉末(过四号筛)200.0 mg 4份,精密称定,分别置具塞锥形瓶中。分别精密加入甲醇10.0、25.0、40.0和50.0 ml,称定重量。分别超声处理(功率250 W,频率40 kHz)30 min,放冷,再按照“2.2.2”项下供试品配制方法配制,即得4份供试品溶液。按照“2.1”项下的色谱条件测定。发现加入10.0 ml溶剂时,色谱峰存在包峰情况;加入40.0和50.0 ml溶剂后测得的含量较加入25.0 ml溶剂测得的含量略有增加,在0.05%左右,故考虑为系统误差。本着节约原则确定提取溶剂体积为25.0 ml,结果见表5。

表5 不同提取溶剂体积考查结果
3.3样品数据分析 本试验将桂枝饮片根据外观形态,如断面直径、皮部颜色、木部颜色等方面区分为三个等级[5]。12批桂枝样品可以分为以下三个品级:A类(批号S3、S6、S11),饮片为类圆形或椭圆形的厚片及段,断面直径均在3~5 mm之间,皮部呈深红棕色,木部呈浅黄棕色,髓部明显略呈方形,特异香气浓郁;B类(批号S2、S5、S9、S10),饮片为类圆形或椭圆形的厚片及段,断面直径在5~10 mm之间,皮部呈红棕色,木部呈浅黄棕色,髓部较A类样品较不明显,略呈方形或类圆形,有特异香气;C类(批号S1、S4、S7、S8、S12),饮片为类圆形或椭圆形的厚片及段,断面直径大小不均匀,大部分断面直径在10 mm以上,皮部呈棕色,木部呈黄白色,髓部较A类样品较不明显,略呈方形或类圆形,有较B类样品不明显的特异香气。如图2所示,各品级桂枝外观形态对比(S11、S2、S8);如图3所示,各品级桂枝横截面对比(S11、S2、S8)。表6列出每批样品的品级、市售价格及测得肉桂酸的含量信息。

图2 A、B、C品级桂枝外观比较

图3 A、B、C品级桂枝横截面比较
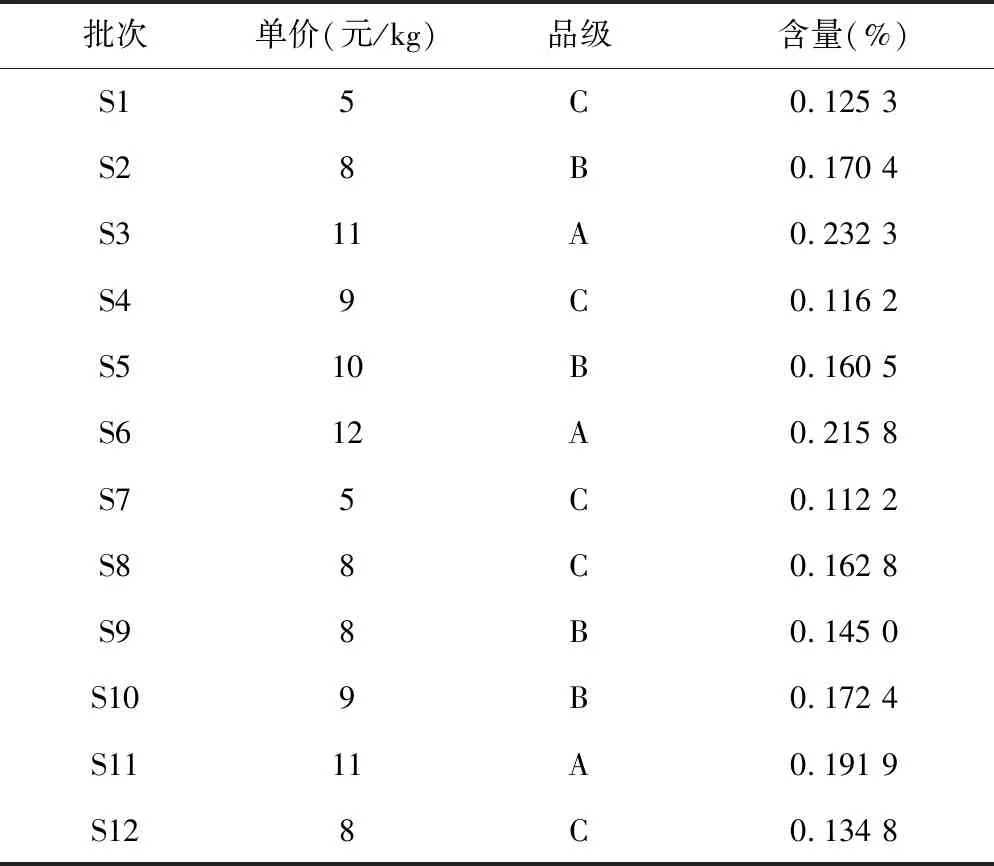
批次单价(元/kg)品级含量(%)S15C0.125 3S28B0.170 4S311A0.232 3S49C0.116 2S510B0.160 5S612A0.215 8S75C0.112 2S88C0.162 8S98B0.145 0S109B0.172 4S1111A0.191 9S128C0.134 8
根据表中信息,绘制价格-含量关系图及品级-含量关系图,如图4和图5。由图4所示,将批号按照市售价格降序排列,折线图代表各批次桂枝价格的次序,柱状图代表每批次桂枝中肉桂酸的含量高低。观察可知,除少数批次(如S4、S5)外,市售价格与肉桂酸含量基本呈正相关。由图5可知,按照批号的品级高低进行排列,折线图代表各批次桂枝价格的高低,柱状图代表每批次桂枝中肉桂酸的含量高低。观察可知,A品级的桂枝明显较B、C品级的桂枝肉桂酸含量高,且价格较高;B品级的桂枝较A品级桂枝在含量方面有所降低,且价格也较A品级低;C品级的桂枝肉桂酸含量参差不齐,但大部分都较A、B品级的低,价格也相差较大。
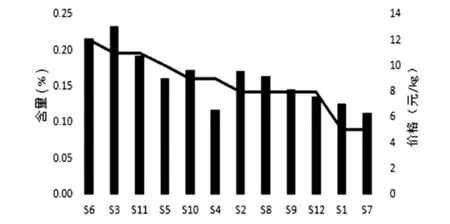
图4 价格-含量关系图
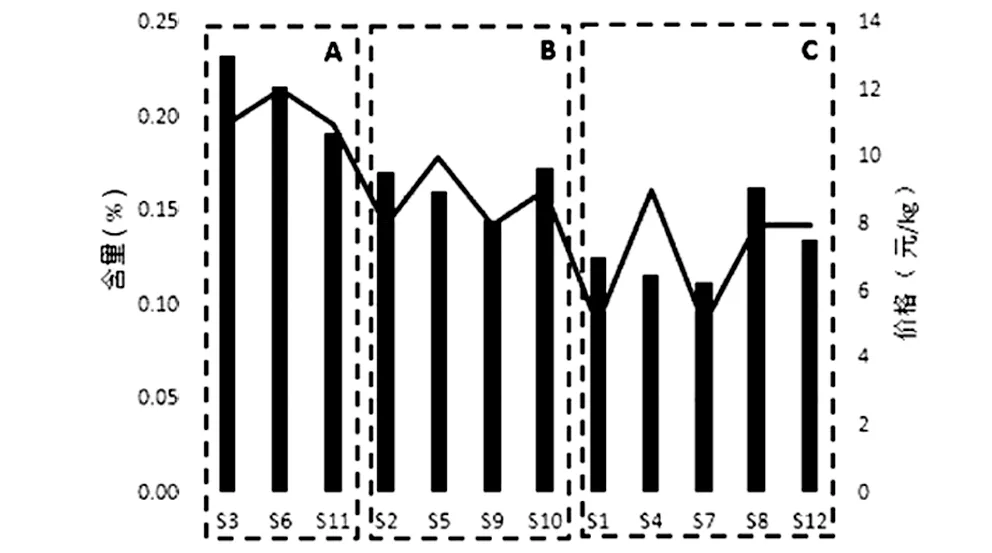
图5 品级-含量关系图
4 结论
中药的质量优劣决定了临床疗效,而且直接关系到人民群众的生命和健康安全,进而影响着中医药事业的发展。由于市场竞争日趋激烈、野生中药材的资源匮乏、中药材需求与产量矛盾加剧[6]等原因,导致中药的质量存在诸多的问题。因此在中药流通市场中找寻一种能够通过外观快速简便地初步识别药品质量优劣的方法就显得尤为重要。
本文建立HPLC法对桂枝中肉桂酸的含量进行测定,该方法具有操作简单、准确度好、灵敏度高等优势,为桂枝提供了质量控制的依据。同时,用该方法对市售12批桂枝药材进行肉桂酸含量测定,从测试结果看来,药材市场上流通的桂枝药材中肉桂酸的含量存在一定差异,且与其外观形态、价格具有一定的相关性。通过数据发现,桂枝饮片的厚片及段的断面直径在3~5 mm之间,皮部呈深红棕色,特异香气非常浓郁的,肉桂酸的含量较高,也就是将品级评为A级的部分;市售价格与品级基本呈正相关,品级较高即枝条较细的桂枝药材肉桂酸含量较高,认为是细枝条中皮部占比较高所致,这与文献报道所述“桂枝皮部肉桂酸含量普遍比木部高”吻合[7]。证明桂枝中肉桂酸含量与桂枝饮片外观形态的相关性较好。研究结果表明,能够通过直观的观察法初步判断桂枝的质量,为市场中初步评价桂枝饮片质量提供便捷有效的方法。
