聚苯撑乙炔螺旋异构体的结构和吸收光谱性质研究
2019-03-29王素凡王慧娟
王素凡, 王慧娟, 许 鹏
(安徽师范大学 化学与材料科学学院,安徽 芜湖 241000)
引 言
含苯环的共轭聚合物由于其良好的光电性、溶解性、易加工性和广泛的应用性而备受青睐[1-3]。主链由亚乙炔基与亚苯基对位交替聚合的线型聚对苯撑乙炔[Poly(p-phenylene ethynylene),p-PPE]聚合物,由于其圆柱状的电子云分布,主链的电子流动性好,共轭长度保持性好,在溶液中有较高的荧光量子效率等独特的光电性能,是可广泛应用于爆炸探测、纳米间隙分子导线、液晶显示起偏器、发光二极管等发光器件和能量传输材料等不同领域的理想材料[4,5]。
在聚苯撑乙炔的光学性能和新材料的开发实验中,人们发现虽然p-PPE具有如上的良好品质,但在不同的合成条件下得到的p-PPE构型不同,温度、溶剂、基质等因素均会影响其构象和光电性质[6-9]。研究发现,对位聚苯撑乙炔的形态取决于其碳骨架的灵活性,通过用四种寡聚物对位聚苯撑乙炔进行自旋标记。在玻璃态溶剂中,改变温度,对位聚苯撑乙炔发生一定的弯曲[10],如果对位聚苯撑乙炔足够长,则可能形成螺旋环状,此特征已经应用于碳纳米管相关性质的研究[11]。聚苯撑乙炔的苯环间位、邻位、邻对位等共聚物的光学活性在生物体系中的应用也是一个非常值得注意的研究方向。由于与生物大分子螺旋状结构的相似性,光学活性螺旋聚合物在合成、结构和功能等多方面引起人们的关注。以聚苯撑乙炔为基元通过自组装设计出各种螺旋结构的聚合物,并且根据其性能开发出一系列新型材料,如:大连理工大学所研究的聚合物材料的荧光传感器[15],日本福井大学对m-PPE的螺旋结构性质的研究[16]以及新加坡研究者对螺旋聚合物与DNA荧光探针的研究[17]等。目前,许多化学家一直在挑战开发人工螺旋聚合物、超分子以及手性低聚物,不仅模仿生物的螺旋结构与功能,也发现其在材料领域的潜在应用[18]。
我们利用自组装方法设计了一系列螺旋环状结构的聚苯撑乙炔异构体,并用密度泛函方法对其结构和性质进行理论计算研究。预测聚合物的微观几何构象、分子结构、共轭链长度等是本质上决定聚苯撑乙炔光学性质的主要因素。聚合物共轭链内在结构特征的理论研究为聚苯撑乙炔的宏观光学性能的改进调制和应用提供了重要的理论依据[19-22]。
1 分子模型设计和理论计算方法

图1 聚苯撑乙炔螺旋结构基元二聚体的形成机理示意图Fig.1 The scheme for the formation mechanism of the helical poly pheneylene ethynylene dimer unit
以苯乙炔基本单元的邻、间、对缩聚异构为基础,利用自组装方法选择苯环不同的聚合位点,设计聚苯撑乙炔二聚体的骨架结构(如图1所示)。其中对位二聚体为直线结构,间位和邻位二聚体的乙炔基间的键角都小于180°,增加聚合单元,对位聚合一般形成链状聚合物,间位和邻位可能形成非直线型的环状或锯齿状立体构型。据此,以邻、间、对聚合位点聚合形成不同的二聚体基元,设计出一系列不同尺寸和拓扑结构的螺旋环状聚合物。在此研究中,为了简化并突出位置异构二聚体的几何特征,用乙炔基取代二聚苯撑乙炔中苯环端基氢原子,可产生对—对位(pp—PPE)、间—间位(mm—PPE)、邻—邻位(oo—PPE)、对—间位(pm—PPE)、对—邻位(po—PPE)、间—邻位(mo—PPE)6种异构化构型,利用其中后5种非线型结构重复聚合来讨论相应的螺旋结构。
运用Gaussian09程序[23]中的密度泛函(DFT)方法在B3LYP/6-31G的水平上对不同的结构模型分子进行基态优化计算和频率计算得到基态热力学稳定几何构型。并利用时间相关的密度泛函方法(TD-DFT),在与基态对应的理论水平下对模型分子的最优结构进行激发态的确认、激发能的计算和吸收光谱的预测,并利用自然跃迁轨道方法分析聚合物的跃迁轨道特征。
2 结果与讨论
2.1 聚苯撑乙炔二聚体基元
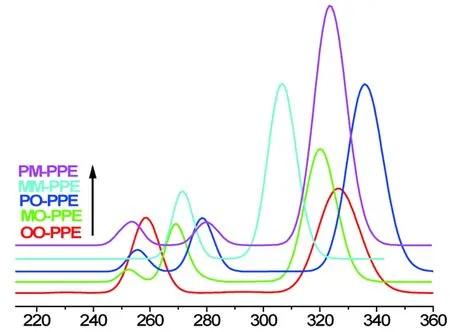
图2 螺旋结构聚苯撑乙炔二聚体基元的吸收光谱示意图Fig.2 The absorption spectra of the helix poly phenylene ethynylene dimmer units
利用密度泛函方法,对乙炔基封端苯乙炔二聚体的异构体进行了几何构型全优化,基态稳定结构几何特征参数如表1,各异构体的热力学稳定性差别很小,可预测均为基态稳定结构。优化得到不同非线型异构体的空间几何结构中苯乙炔共轭面间的二面角均为0度,说明二聚体均为平面型共轭分子。具体的成键特征为,单体聚合方式对苯环间的乙炔基碳碳三键影响不明显,键长均约为1.220Å,而乙炔基与苯环之间的碳碳单键键长区别较大,最小的是邻—邻位与邻—对位异构体,键长约为1.426Å,最长的是间—对位结构,键长为1.428 Å。我们发现有邻位聚合时,键长都偏短,含间位时,键长偏长,可以预测在含有邻位作用的二聚体结构中,乙炔基的碳原子可与苯环产生较大的离域共轭作用。邻位的键角较小,从几何形貌看,类似正三角形,邻—邻位键角为67.84度,邻—对位键角为62.24度,而邻—间为有两个键角,邻位的为62.14度,其键角大小的区别主要是因为其端基的影响,邻—邻位两乙炔基空间位置近,氢原子之间的排斥力较大,所以导致键角是邻位中最大的一个,而邻—间位、邻—对位排斥作用依次减小,所以键角也相应的次序减小,间位的左右作用特征相似,邻—间位中的间位键角最大,间—对位最小。
基于基态稳定几何构型,利用时间相关的密度泛函方法(TD-DFT)对聚苯撑乙炔基元二聚体异构体的激发态性质进行理论计算,得到对应的吸收光谱如图2所示,五种异构体具有相似的吸收特征峰,吸收光谱的范围从296nm到331nm,并且随着聚合位点的不同,最强吸收峰的位置和共振强度均有一定的差别。其中po—PPE具有最长吸收谱线,而pm—PPE具有较强的共振强度。各种异构体的最强激发态跃迁参数列于表2,可以看出异构体的最强跃迁均为第一激发态前线轨道HOMOLUMO的跃迁,oo—PPE、om—PPE、mm—PPE激发能逐渐增大,其波长相应变短,而含对位的二聚体激发能均偏小,其中以po—PPE激发能最小,吸收波长最长,可预测其共轭程度最大。pm—PPE异构体具有最大的跃迁共振强度,吸收光谱的性能最好。基于五种二聚体异构体的光谱性质,可根据共聚位点的差别设计得到具有不同特征吸收的光学材料,此计算结果可为较大分子量的聚苯撑乙炔高聚物在光学器件中光谱性质的可调制性提供一定的理论基础。

表1 螺旋结构聚苯撑乙炔二聚体基元形貌和特征几何参数**Table 1.The pattern and the characteristic structural parameters for the helical poly pheneylene ethynylene dimer units

表2 聚苯撑乙炔二聚体强激发态的跃迁参数Table 2 The parameters for the intensive excitation of the poly pheneylene ethynylene dimers
聚苯撑乙炔二聚体各异构体的最强跃迁均为第一激发态的HOMOLUMO轨道跃迁。在HOMO轨道中,成键作用主要分布在两个乙炔基之间以及部分苯环上,而乙炔基与苯环主要是反键作用。当电子被激发跃迁到LUMO轨道时,电荷布居恰好与HOMO轨道相反,对应反键轨道变为成键轨道,成键轨道变为反键轨道,由此可以预测异构体的特征跃迁均为共轭分子的局域电子的ππ*跃迁。但五种二聚体跃迁后的电荷分布,跃迁轨道能和相应的组成随着异构体的共聚位点的变化,产生了一定的差别。oo—PPE和mm—PPE的电荷为对称分布,乙炔端基的电荷布居较少,电荷布居主要分布在两个苯环和中间亚乙炔基上;po—PPE和pm—PPE的轨道电荷布居相似,其邻位和间位端基的电荷布居较少,对位的电荷分布较为密集,整个分子轨道的电荷主要来源于对位片段的贡献,而om—PPE的乙炔端基电荷分布很少,主要是两个苯环之间的电荷布居较为密集。由轨道上的电荷布居可以看出po—PPE和pm—PPE的异构体中的直链共轭链长主要为两个完整的苯乙炔基元,而其它异构体的电荷布居主要集中在苯环之间,对应的共轭链长较短,由此微观电子结构特征可以说明含有对位片段的异构体由于其较大的离域共轭程度,跃迁能低,吸收波长较长;含有间位基元异构体的共轭程度一般都较小,对应吸收波长较短。
2.2 聚苯撑乙炔基元螺旋结构
以二聚体为结构单元进一步聚合可设计并优化得到不同形貌的单螺环结构苯乙炔聚合物如表3,五种螺旋结构基元基本几何形貌为三角形、四边行以及六边形等类多边形。从俯视图的角度观察聚苯撑乙炔的形貌,发现其类似闭环结构,说明此结构具有较大的成环可能性。在不同的基元螺旋结构中,乙炔基碳碳三键的键长与其二聚体结构基元相似没有发生改变。乙炔基与苯环碳原子之间键长发生一定改变,其单键键长大小顺序为om—PPE最大,然后是mm—PPE、pm—PPE、oo—PPE、po—PPE依次减小。相应基元结构的键角也有一定差别,mm—PPE的键角较大,其中pm—PPE的键角为120.84度,mm—PPE的键角为121.27度,po—PPE较为特殊,其间位键角为137.00度,而邻位的键角一般较小,其键角为80.00度。邻—邻位的键角为68.68度,邻—对位的键角最小为67.55度。从聚苯撑乙炔螺旋形貌看出,基元螺旋结构具有比较紧凑的形貌,整个螺环电子云分布较为密集,原子间存在较大的排斥作用,使得螺旋结构的键长、键角发生相应的改变。特别是键角和二面角的变化,与二聚体的几何参数相比,含有邻位结构的螺环基元键角明显增大,其可能原因是四边形和三角形螺环基元空间环较小,骨架内侧原子间具有更大的空间位阻。二面角的产生主要是由于螺旋基元首尾端基的相互影响,从表3中的边长对比可以看出端基一侧可形成约0.7Å的重叠,由于空间位阻和原子间的排斥作用使得聚苯撑乙炔间二面角发生偏转,形成螺旋结构。

表3 聚苯撑乙炔聚合物基元螺旋结构的形貌和特征几何参数**Table 3 The pattern and the characteristic structural parameters for the single helical PPE polymers
**All the data of bond length and angle are the average value for the whole structure

图3 聚苯撑乙炔聚合物基元螺旋结构的吸收光谱示意图Fig.3 The absorption spectra of the single helical poly phenylene ethynelene polymers
利用时间相关的密度泛函方法,计算聚苯撑乙炔螺旋结构基元的激发态性质,理论预测的吸收光谱如图3,螺旋结构基元与二聚体的光谱性质有明显的差别,第一激发态的长波吸收峰(max)的波长发生不同程度红移,强度变弱。强吸收系数的特征强吸收(stro)为高激发态吸收,对应的吸收谱线位置与二聚体的强吸收比较分别发生红移或蓝移。具体的跃迁参数和轨道特征参数列于表4,对于第一激发态的长波吸收均为HOMOLUMO轨道跃迁,特征吸收一般为HOMO-1LUMO,LUMO+1的跃迁(mm—PPE为HOMOLUMO+3)。
特征的强吸收中,pm—PPE和po—PPE的吸收光谱发生了明显的红移,红移的波长分别为54.34nm和34.46nm;mm—PPE的波长只有2.48nm的红移,但om—PPE和oo—PPE却是发生了蓝移,波长分别为5.49nm和39.59nm。由此可以初步预测此种变化的原因可能是因为螺旋聚苯撑乙炔的有机体系的共轭程度发生了明显的变化。首先的主要原因是在螺旋结构中所含有的二聚体基元数目不同,其中pm—PPE的可含有6个基元,使其整体结构的共轭程度明显增大,产生明显红移。而对于mm—PPE和po—PPE来说,二者都包含了3个二聚体基元,但红移的程度却又有很大差别,可以预测增加重复单元对po—PPE体系的电子离域共轭的促进作用较大。对于om—PPE来说,虽然增加了1个重复单元,但从几何形貌上可以看出此时却在螺旋结构中同时产生了较大的二面角,对整个体系的电子离域共轭程度有较大影响,影响其吸收光谱。在oo—PPE的螺旋结构比二聚体增加一个苯环,但是由于强烈的空间位阻效应(二面角为对应体系最大!),破坏了原有电子离域共轭效应而发生较强的吸收光谱蓝移。由此可得,在聚苯撑乙炔聚合物中直接影响体系光学性质的内在因素不完全是体系分子链的长短,更主要是电子离域共轭效应程度的大小,此结果与相关体系的研究相吻合[24]。单层螺旋结构聚合物的吸收光谱范围286nm到368nm(如图3),光谱谱线间有82nm的差别,更加利于实验研究的检测与分析,说明这种结构的螺旋聚合物在材料的开发中有良好前景。
与二聚体的HOMO到LUMO轨道跃迁为最强吸收不同,螺旋聚合物的跃迁轨道组成更为复杂,前线轨道中的占据轨道能级发生一定变化,螺旋聚合物前线轨道跃迁时,大部分主要是HOMO-1轨道跃迁,相应的跃迁空轨道之间存在明显简并现象。如表4中各螺旋聚合物的轨道跃迁示意图,可发现跃迁后的轨道电子布局特点。mm—PPE和oo—PPE和po—PPE的跃迁轨道中电子均呈对称性分布,mp—PPE跃迁轨道的电子分布在中间两苯环及乙炔基上或是均分在首尾中三部分,而om—PPE比较特殊,其电子分布主要是集中在螺旋环状的中部和含有乙炔基的端基部分。与相应的二聚体轨道电荷布居相比,pm—PPE和po—PPE结构中的跃迁轨道片段共轭范围明显增大,特别是pm—PPE中,主要布局的共轭增大了接近一个二聚体单元,所以由此可以更直观地解释此结构产生较大红移的微观原因。而对于oo—PPE强烈蓝移的结构中,由于较大的平面畸变,破坏了电子布居的微观对称性,苯环与乙炔基之间的轨道重叠减弱,致使体系的跃迁能增大,吸收光谱蓝移。电子布居直接影响螺旋聚合物的稳定性和几何参数,也从微观方面为其光谱性质理论分析提供了基础。
2.3 聚苯撑乙炔螺旋结构光谱性质
基于单层基元螺旋结构,利用相同的理论和方法构建并计算了两层和三层螺旋结构聚苯撑乙炔聚合物,螺旋聚合物光谱性质和电子跃迁轨道随着聚合物分子量的增加所呈现不同的变化特征。各个螺旋结构的特征跃迁性质与参数列于表5中,与单层基元螺旋结构的跃迁特征相比,随着螺旋层数的增加,对应的第一激发态的最长吸收波长(S1态对应的吸收波长)和最强吸收的波长及其共振强度变化各不相同。总体来说随着螺旋层数的增加,除pm-PPE外,聚合物的对应吸收谱线均一定程度红移,其中po-PPE的红移程度最明显,可达到100nm。
随着螺旋层数的增加,对应特征跃迁的活性轨道能量和电荷布居特征均有一定的变化,出现多重简并轨道的特征,如表6所列的HOMO和LUMO轨道电荷布居随螺旋层数变化出现局域分布的特征,在多层结构中,轨道间的π电子耦合作用使得HOMO轨道能级一致性地升高,其中含有对位单元的pm-或po-构型中HOMO轨道能级变化相对较小,最低约为0.06eV,含有邻位单元的oo-或om-构型中HOMO能级升高较大,最高约为0.48eV。其能量的变化在一定程度上反映出层状结构中的层间作用特征,由几何优化的数据可以得出含有对位结构单元的层间距离较小,而含有邻位结构单元的层间距较大。与HOMO的能级变化相比,LUMO的能级变化较小,一般在0.01—0.04eV之间。由此可以预测多层结构的跃迁主要会受到层间电荷耦合作用的影响,需更进一步的研究来分析不同结构的耦合作用贡献。
3 结论与展望
通过自组装方法设计了一系列聚苯撑乙炔螺旋结构聚合物,运用密度泛函方法对几何结构和光谱性质以及跃迁的分析,确认了各种螺旋结构聚合物的可能光谱范围,影响光谱性质的主要因素,以及轨道跃迁机理。从基元二聚体到螺旋结构光谱性质的变化能够为相应的特征螺旋结构光学材料的实验开发提供坚实的理论基础。尤其在生物材料领域上,聚苯撑乙炔具有良好的导电性能和非线性光学性能,因此模仿DNA螺旋结构的聚苯撑乙炔可以做为生物检测中的荧光传感器材料,与生命科学研究相结合将是聚苯撑乙炔在应用上的一项重大突破。

表4 聚苯撑乙炔聚合物基元螺旋结构最长和最强激发态的跃迁参数Table 4 The parameters for the first and intensive excitation of the single helical PPE polymers

表5 聚苯撑乙炔聚合物多层螺旋结构特征跃迁参数Table 5 The characteristic excitation parameters of the multiple helical layer PPE polymers

表6 聚苯撑乙炔聚合物多层螺旋结构HOMO和LUMO轨道结构和能量Table 6 The HOMO and LUMO sketch and ortibal energy (in eV)of the multiple helical layer PPE polymers
续表6

LayerOrbitalpm-PPEmm-PPEom-PPEoo-PPEpo-PPE2HOMOLUMO-5.45-5.61-5.49-5.45-5.29-1.98-1.59-1.64-1.58-2.083HOMOLUMO-5.46-5.51-5.33-5.10-5.20-1.94-1.55-1.69-1.62-2.03
