利用CRISPR/Cas9基因编辑技术靶向敲除奶山羊胎儿成纤维细胞SCD1基因
2019-03-26刘雷雷贾启鹏李宗帅杨洋李海江赵兴绪张勇
刘雷雷,贾启鹏,李宗帅,杨洋,李海江,赵兴绪,张勇
(甘肃农业大学动物医学院,甘肃 兰州 730070)
硬脂酰辅酶A去饱和酶1(stearoyl-CoA desaturase-1,SCD1)是1种存在于内质网的内膜蛋白[1],在合成单不饱和脂肪酸(MUFA)棕榈油酸和油酸的过程中具有重要作用[2],可以将饱和脂肪酸分解为不饱和脂肪酸,这一酶促反应在机体的不饱和脂肪酸储备中起到重要作用[3].同时SCD1还能催化trans11-C18∶1(tll-C18∶1)生成cis9,trans11-共扼亚油酸(c9,t11-CLA)[4].MUFA和共扼亚油酸(CLA)具有调节血液中胆固醇含量,防止心血管疾病,提高免疫力等作用,其中CLA能够有效抑制肿瘤生长[5].因此,通过对敲除SCD1基因动物模型的研究,可以间接研究人类一些疾病的发病机制[6].
基于转基因动物平台的动物乳腺生物反应器,是在动物基因组中通过转入外源性靶基因并定位表达于乳腺,利用其天然、高效分泌蛋白的能力,在动物的乳汁中生产一些具有重要价值产品的转基因动物的总称[7].有研究报道乳制品提供高质量的蛋白质和其他微量营养素,对人类饮食中不饱和脂肪酸有显著贡献[8],而不饱和脂肪酸的含量与人类许多疾病有着密切关系,因此,可以使用基因编辑技术来增加动物乳汁中不饱和脂肪酸含量,从而来改善人类的身体健康.牛乳中含有丰富的s1-酪蛋白,它是人体对乳蛋白最主要的过敏源,然而,羊奶在营养成分上更接近于人奶,其富含β-乳球蛋白,赋予其更高的可消化性,并降低了其易过敏性,更适合老年人和婴幼儿等肠胃较为脆弱的人群饮用[9],因此,可以通过基因编辑技术改良山羊品种从而提高羊奶品质.
CRISPR/Cas9系统是由规律成簇的间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)结构与一些功能相关的蛋白(CRISPR-as-sociated,Cas)组成的一种定点基因编辑技术,当其被发现后科学家已证实该技术在细菌[10]、植物[11]、动物[12]以及人类细胞[13]上都表现出强大的基因编辑能力.由于CRISPR/Cas9系统拥有简单、高效的作用机制[14],使其应用于转基因动物平台来生产动物模型,研究人类疾病或改良家畜品种及其衍生产品营养价值,必然会成为研究热点[15-17].本试验利用CRISPR/Cas9系统在奶山羊胎儿成纤维细胞SCD1基因设计并筛选出能够高效切割的sgRNA,来证实该基因编辑技术可以应用于奶山羊,为以后在该动物敲入外源性基因奠定理论基础;同时使用双敲除载体共转染奶山羊胎儿成纤维细胞的方法来获得敲除SCD1基因的GFFs,为生产敲除SCD1基因奶山羊动物模型提供核供体材料.
1 材料与方法
1.1 试验材料
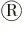
奶山羊胎儿成纤维细胞F2代,实验室保存;PX330-eGFP质粒,实验室保存;pEASY-Blunt载体购自北京全式金生物技术有限公司.
1.2 试验方法
1.2.1 引物设计 如表1所示,使用Primer5设计特异性检测引物后,送金唯智公司合成.

表1 检测引物序列信息
1.2.2 sgRNA的合成及程序性退火 通过查找奶山羊SCD1基因编码区序列(ID:GU947654.1),使用靶位点在线预测工具(http://crispr.mit.edu/),按照“20N+NGG”原则在该基因第4外显子和第6外显子处设计6对sgRNA寡聚核苷酸序列(表2),分别在有义链5′端添加CACC,模板链5′端添加AAAC酶切位点送金唯智公司合成之后,将单链sgRNA程序性退火变为含黏性末端的双链.
1.2.3 敲除载体的构建 使用限制性内切酶BbsⅠ对PX330-eGFP质粒进行酶切处理后,纯化回收酶切产物.然后,利用T4DNA连接酶将程序性退火的双链sgRNA连接到酶切后纯化回收的产物,之后转化到Trans1-T1化学感受态细胞中,并均匀铺于含氨苄抗性的LB固体培养皿上37 ℃过夜培养,第2天挑取单菌落溶于10 μL ddH2O,以U6上游引物和各个sgRNA的oligo2为下游引物进行菌液PCR及测序验证.

表2 设计的sgRNA寡聚核苷酸序列
1.2.4 细胞的转染及切割效率的检测 复苏奶山羊胎儿成纤维细胞F2代,经传代培养细胞汇合度达到70%~80%时,按照LipofectamineTM2000转染试剂盒说明书进行转染,待转染48 h后使用流式细胞分选仪分选出绿色荧光细胞并提取细胞基因组DNA,之后使用高保真DNA聚合酶扩增各靶点上下游共计609 bp(上游引物为J4F,下游引物为J4R)及703 bp(上游引物为J6F,下游引物为J6R)的片段,经1%的琼脂糖凝胶电泳检测后回收目的片段.将目的片段连接到pEASY-Blunt载体,转化到Trans1-T1化学感受态细胞中,涂布于氨苄平板上37 ℃过夜培养,次日挑取单菌落,PCR检测(上游引物为M13F,下游引物为M13R)后送生物公司测序.
1.2.5 PX330-eGFP-sgRNA共转染细胞 分别选择第4外显子和第6外显子处具有高效敲除效率的sgRNA两两组合,按照4 μg质粒和10 μL脂质体共同转染到奶山羊胎儿成纤维细胞中,48 h后使用流式细胞分选仪筛选出绿色荧光细胞继续培养.
1.2.7 Western Blot检测 提取绿色荧光细胞总蛋白后,步骤参照文献报道的方法进行[18].相应的一抗(按1∶400 的比例溶解于5%脱脂奶粉的PBST溶液)4 ℃过夜孵育,辣根过氧化物酶标记相应的二抗(按1∶5000 的比例溶解于5%脱脂奶粉的PBST溶液)37 ℃孵育2 h,加入化学发光液后,X胶片曝光进行显影和拍照.
M:DNA marker DL10000;1:PX330酶切;2:PX330质粒.M:DNA marker DL10000;1:PX330 digestion;2:pX330 plasmid.图1 PX330-eGFP酶切鉴定Figure 1 The identification results of PX330-eGFP digestion
2 结果与分析
2.1 敲除载体的构建
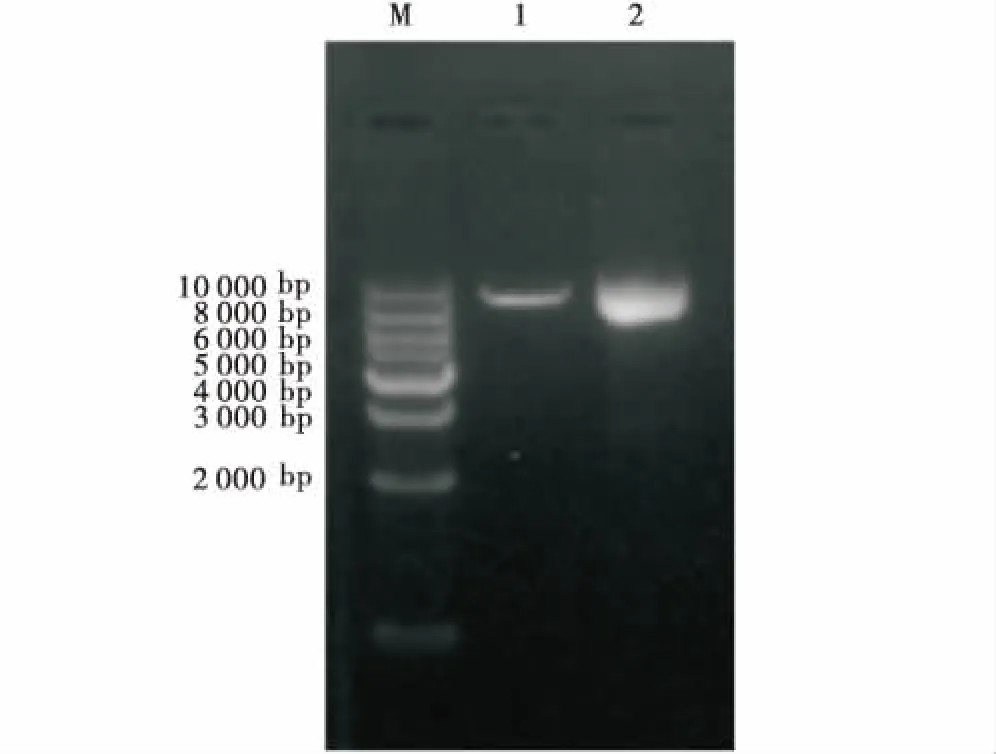
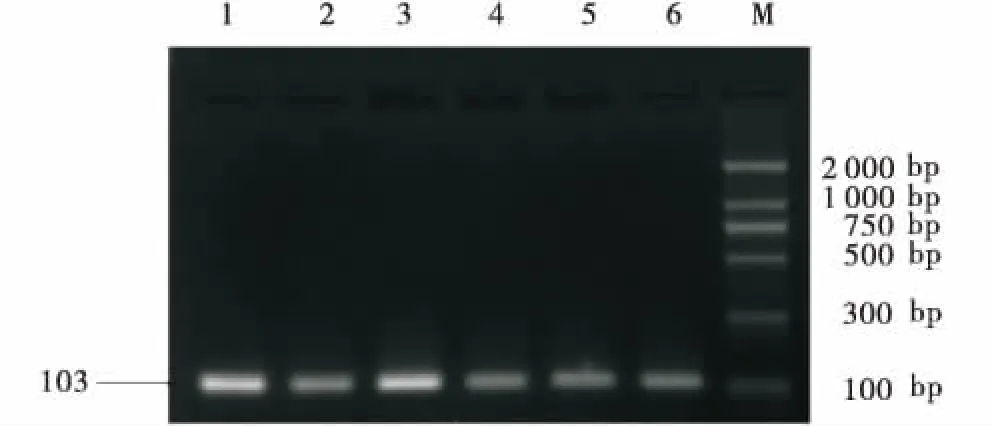
如图1所示,PX330-eGFP经限制性内切酶BbsⅠ单酶切后出现与预期结果大小相符的条带,表明PX330-eGFP质粒酶切成功.使用U6上游引物和各个sgRNA的oligo2为下游引物挑取阳性菌落,如图2所示,菌落PCR琼脂糖电泳结果与目的条带(103 bp)大小相符.接着将条带大小相符的菌落摇菌扩大培养,次日提取质粒后送生物公司测序.如图3所示,测序峰图表明PX330-eGFP-sgRNA敲除载体构建成功.
M:DNA marker DL2000;1:sgRNA F1;2:sgRNA F2;3:sgRNA F3;4:sgRNA S1;5:sgRNA S2;6:sgRNA S3.M:DNA marker DL2000;1:sgRNA F1;2:sgRNA F2;3:sgRNA F3;4:sgRNA S1;5:sgRNA S2;6:sgRNA S3.图2 Oligo退火产物连接至PX330-eGFP菌落PCR结果Figure 2 PCR result of the oligo plus PX330-eGFP plasmid construct
图3 Oligo退火产物连接至PX330-eGFP质粒测序峰Figure 3 Oligo annealing product is ligated to PX330-eGFP plasmid sequencing peak
2.2 奶山羊胎儿成纤维细胞的转染结果
如图4所示,PX330-eGFP-sgRNA载体转染奶山羊胎儿成纤维细胞48 h后,均出现绿色荧光阳性细胞,表明奶山羊胎儿成纤维细胞转染成功.
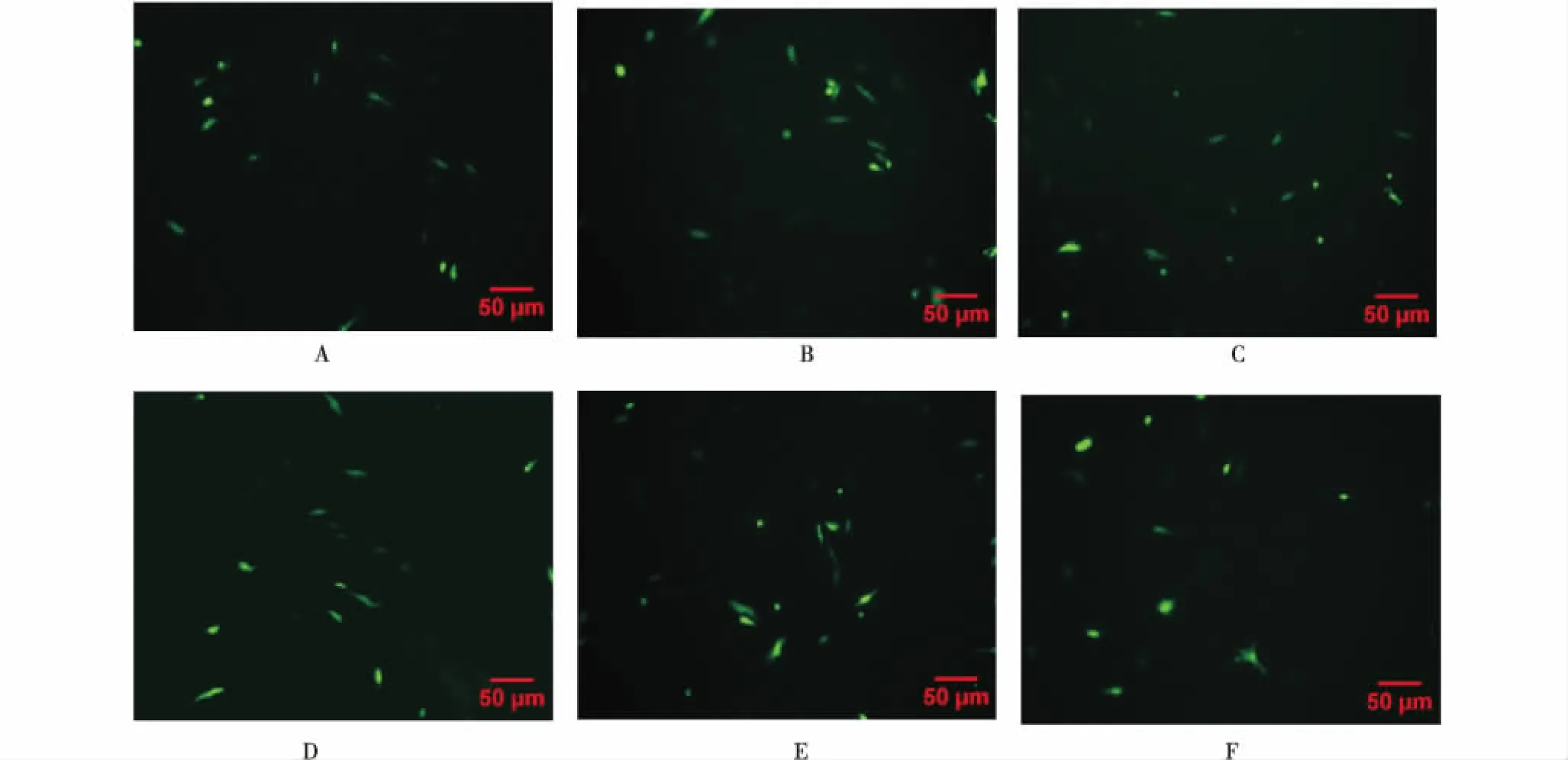
A-F分别是敲除载体PX330-eGFP-sgRNAF1、PX330-eGFP-sgRNAF2、PX330-eGFP-sgRNAF3、PX330-eGFP-sgRNAS1、PX330-eGFP-sgRNAS2、PX330-eGFP-sgRNAS3转染GFFs 48 h后的结果.A-F were the result of knockout vectors PX330-eGFP-sgRNAF1,PX330-eGFP-sgRNAF2,PX330-eGFP-sgRNAF3,PX330-eGFP-sgRNAS1,PX330-eGFP-sgRNAS2,and PX330-eGFP-sgRNAS3 were transfected into GFFs for 48 h.图4 奶山羊胎儿成纤维细胞转染48 h荧光表达(X100)Figure 4 Fluorescent expression of fetal goat fibroblasts transfected for 48 h(X100)
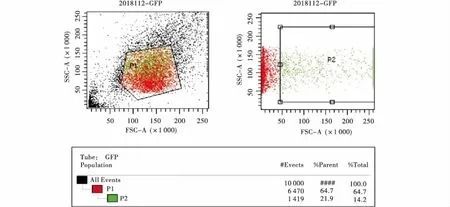
图5 荧光细胞流式分选结果(示部分)Figure 5 The results of fluorescent cell flow sorting
2.3 分选阳性细胞及其PCR检测
使用流式细胞仪在515 nm蓝光波长激发下分选出荧光细胞,如图5所示,荧光细胞转染率可以达到14.2%.提取荧光细胞基因组DNA,使用检测引物J4F/J4R和J6F/J6R分别对阳性细胞进行PCR扩增,如图6所示,得到条带大小与预期条带大小(609、703 bp)相符合,说明阳性细胞扩增成功.
2.4 不同靶位点sgRNA敲除效率的筛选
将TA克隆菌落PCR DNA片段检测阳性的质粒送公司测序后,使用DNAMAN软件将测序结果和野生型(WT)序列进行比对,结果如图7所示(示部分结果).根据DNAMAN比对结果对每个sgRNA在靶位点造成的突变、插入或删除情况进行统计,如表3所示,结果表明6个sgRNA中sgRNA F1与sgRNA S3发生脱靶, sgRNA F2、sgRNA F3、sgRNA S1和sgRNA S2具有敲除作用,其效率分别为41.67%、30.77%、6.67%和28.57%.
2.5 敲除SCD1基因GFFs的制备
将试验组和对照组转染奶山羊胎儿成纤维细胞,待48 h后分选荧光细胞,提取细胞中总RNA和总蛋白.使用实时荧光定量PCR方法检测细胞SCD1在mRNA水平上的变化情况.使用Western Blot方法检测细胞SCD1在蛋白水平上的表达.将实时荧光定量结果使用Tukey法比对分析后,结果表明双载体敲除细胞的SCD1在mRNA水平上的表达与对照组相比存在显著差异(图8,P<0.05),sgRNA-F2-S2与sgRNA-F3-S2不存在显著差异,说明两种组合都可以有效降低奶山羊胎儿成纤维细胞SCD1在mRNA水平上的表达.Western Blot结果显示,在双载体转染的阳性荧光细胞中未表达SCD1蛋白(图9),说明sgRNA-F2-S2与sgRNA-F3-S2 2种组合均能有效敲除奶山羊胎儿成纤维细胞中SCD1基因.
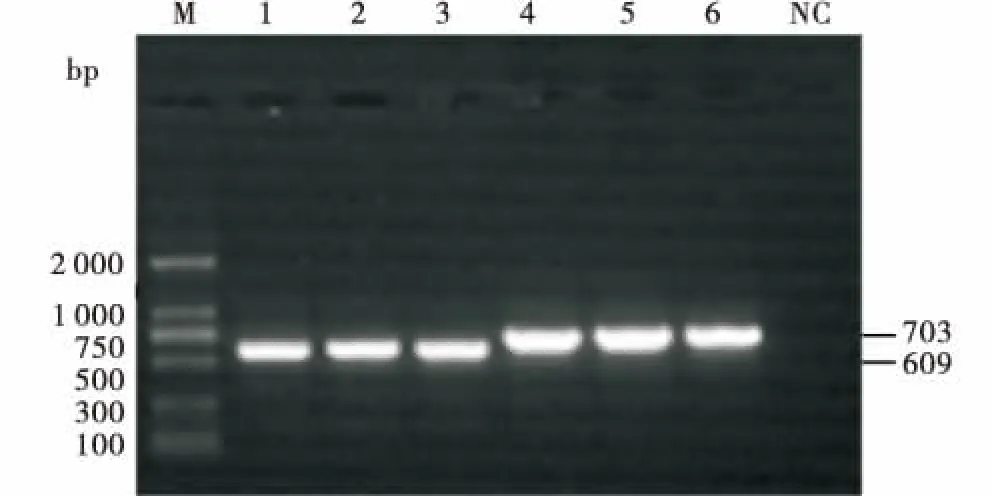
M:DNA marker DL2000;1:sgRNA F1;2:sgRNA F2;3:sgRNA F3;4:sgRNA S1;5:sgRNA S2;6:sgRNA S3;NC:阴性对照.M:DNA marker DL2000;1:sgRNA F1 test result;2:sgRNA F2 test result;3:sgRNA F3 test result;4:sgRNA S1 test result;5:sgRNA S2 test result;6:sgRNA S3 test result;NC:Negative control.图6 荧光细胞基因组DNA PCR电泳Figure 6 Agarose electrophoresis of PCR products from fluorescent cell genomic DNA
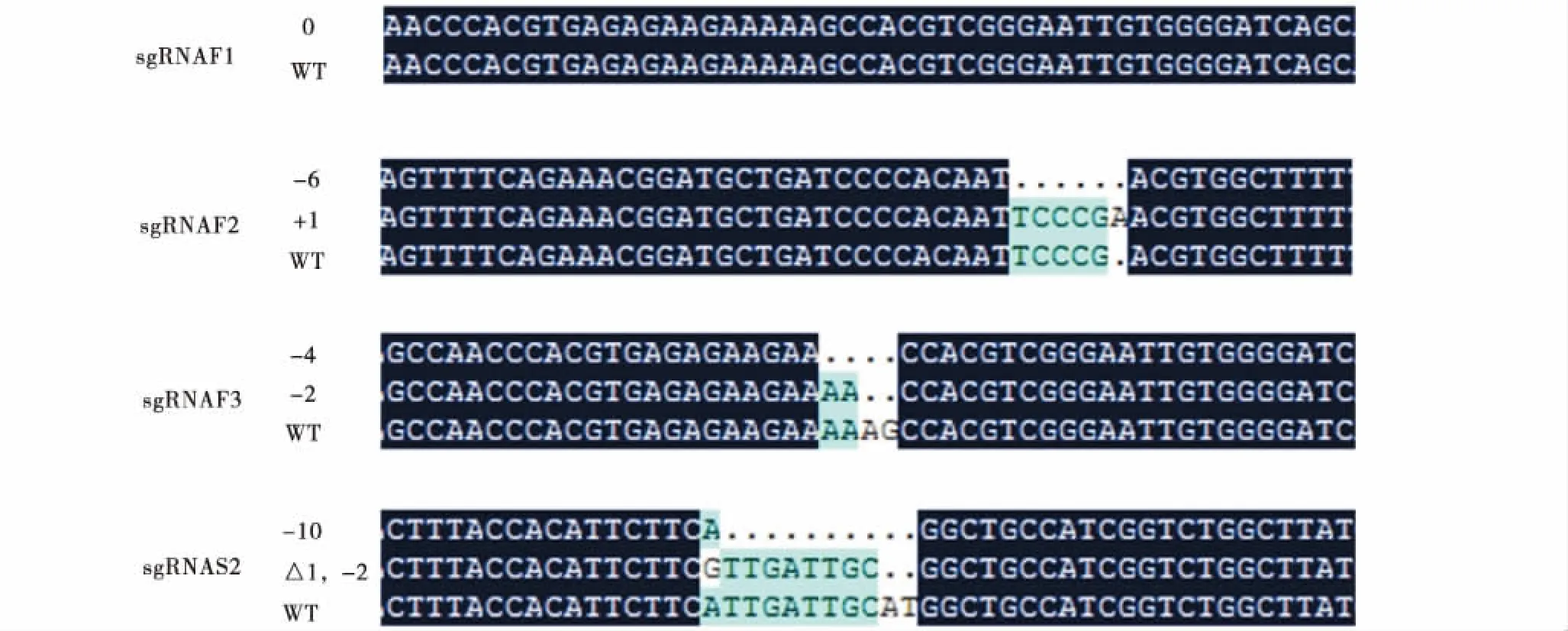
-:碱基删除;+:碱基插入;△:碱基突变.-:Base deletion;+:Base insertion;△:Base mutation.图7 靶位点PCR DNA片段TA克隆测序比对结果(示部分)Figure 7 The results of target site PCR DNA fragment TA cloning sequencing alignment genome
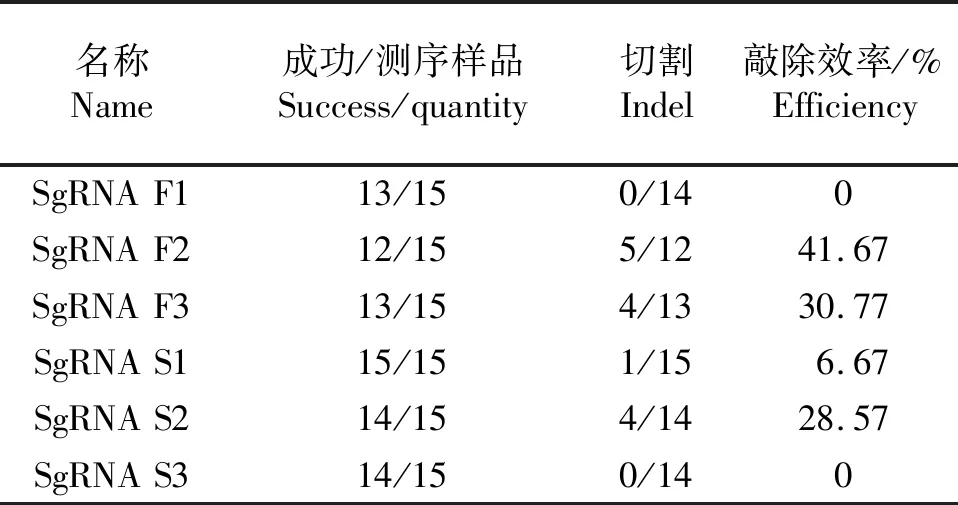
表3 靶位点sgRNA敲除效率统计Table 3 Knockout efficiency of sgRNA at target site
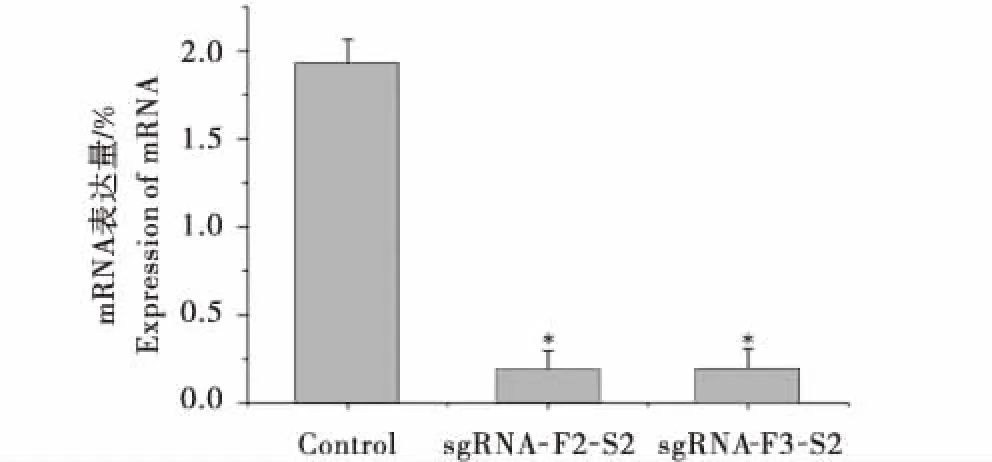
图8 各组细胞中SCD1基因mRNA的表达Figure 8 Expression of SCD1 mRNA in each group of cells
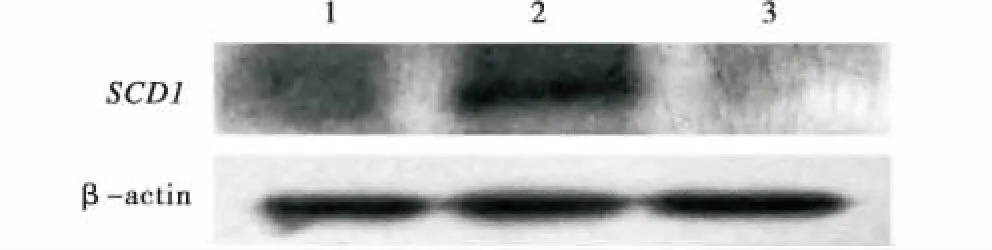
1:转染sgRNA-F2-S2阳性细胞总蛋白;2:GFFs总蛋白;3:转染sgRNA-F3-S2阳性细胞总蛋白.1:Total protein transfected with sgRNA-F2-S2 positive cells;2:Total protein of GFFs;3:Total protein transfected with sgRNA-F3-S2 positive cells.图9 Western Blot检测SCD1蛋白表达Figure 9 Detection of SCD1 protein expression by Western Blot
3 讨论
CRISPR/Cas9系统提供了简单的,比ZFNs和TALENs更吸引人的基因编辑靶位点的选择方法[19],其核酸酶靶向编辑基因组的特异性体现于sgRNA中20个碱基的间隔序列(spacer)与基因组中的靶基因互补识别,从而介导Cas9核酸酶在靶点处切割DNA,产生双链切口,实现基因的定点修饰[20].所以,CRISPR/Cas9介导的SCD1基因敲除载体的构建实质是将20 bp靶序列DNA克隆至2段正向重复序列之间.本试验设通过设计2条互补的寡聚核苷酸直接程序性退火产生两端带有酶切位点靶序列DNA,然后将其克隆至含有相同酶切位点的PX330-eGFP载体,获得针对敲除SCD1基因的载体,该方法设计简易、操作难度小以及成本低,可以实现CRISPR/Cas9敲除载体的快速构建.
脱靶现象几乎是所有基因定点编辑技术所面临的1个主要问题.CRISPR/Cas9技术因操作简易、高效、成本低等优势[21],已被成功应用于小鼠、斑马鱼、果蝇及大动物等多种模式动物,包括基因功能研究[22]、遗传疾病治疗等研究[23].然而其较高的脱靶效应也受到了极大的关注,如若发生脱靶会造成基因组不稳定,正常基因功能的丧失,限制了其在治疗和临床医学中的应用[24].有研究显示该系统在发挥基因编辑功能时造成脱靶效应原因主要与sgRNA临近PAM序列的GC含量有关[25].当选择1个合适的sgRNA 时,Guide序列第1个碱基处鸟嘌呤(G)是必须首选的,而胞嘧啶(C)是坚决避免的.相反,在PAM序列附近第5个碱基处,首选的是胞嘧啶(C),而不是鸟嘌呤(G)[26].本试验设计了6个sgRNA,通过对阳性细胞靶位点的DNA片段进行TA克隆和测序分析,发现sgRNA F2、sgRNA F3、sgRNA S1及sgRNA S2切割效率分别为41.67%、30.77%、6.67%、28.57%,而sgRNA F1和sgRNA S3未发生基因编辑作用.其中sgRNA F2切割效率最高是由于设计的Guide序列第1个碱基为G,且G的含量较其它序列丰富,在体内可以折叠形成G-四联体,对sgRNA的稳定性有一定的作用,但是sgRNA F1和sgRNA S3发生脱靶分析原因是由于Guide序列第1个碱基不是G,并且临近PAM序列附近第5个碱基为G.因此,如何在提高Cas9切割效率的同时,减小脱靶效应是CRISPR/Cas9开展应用要进一步研究和解决的科学问题.目前的研究主要集中在sgRNA的选择、Cas9 的改造等减少脱靶效应的实验技术方面.第一,通过优化设计sgRNA的寡聚核苷酸序列,如在其3′端减去一些核苷酸序列或者在其5′端添加个别序列,来改变sgRNA与靶位点互补的序列[27].第二,通过控制转染时sgRNA的用量,有研究表明转染时低剂量的Cas9-sgRNA复合物浓度可以有效降低脱靶效率,但是过度降低sgRNA的用量也会导致其敲除效率的下降,所以需要合理的控制增加靶位点敲除效率与降低脱靶效应之间的关系[28].第三,通过改造Cas9核酸酶,在Cas9的2种结构域位点RuvC或NHN分别插入D10A或H840A的突变,形成只切割靶位点1条链的Cas9突变酶,并应用1对sgRNA分别在单链DNA上进行切割,来降低脱靶效应[29].第四,重新组装无活性的Cas9与Fok I,有研究显示将无活性的Cas9与Fok I 重新组装形成f-Cas9复合物,其切割靶序列的特异性高出野生型140倍,并且比突变Cas9核酸酶的特异性也高出4倍[30].
由于设计的sgRNA靶向SCD1基因的编码序列进行切割,因此很大程度上能影响其蛋白的表达.一般真核生物体内断裂的DNA双链缺口是通过同源重组(homologous recombination,HR)和非同源末端连接(non-homologous end joining,NHEJ)2种方式来进行修复.HR修复可以将外源基因敲入到靶位点修复断裂的DNA双链,来生产转基因动物.而NHEJ修复断裂的DNA双链,是通过引发切割位点DNA碱基插入、缺失或替换,从而实现目的基因的敲除[31].本研究就是利用NHEJ方式来修复DNA双链缺口,同时还发现各sgRNA引起靶点部分碱变化数大多为3的非整数倍,CRISPR/Cas9系统是通过移码突变造成基因组蛋白无法正常被翻译,从而来实现SCD1基因真正意义上的敲除[32].
4 结论
本研究利用CRISPR/Cas9系统成功从奶山羊胎儿成纤维细胞SCD1基因第4外显子和第6外显子筛选出高效切割的sgRNA,其切割率最高分别为41.67%和28.57%,验证了该技术在奶山羊基因编辑应用中的有效性.使用双载体转染方案,获得了敲除SCD1基因的GFFs,为制备敲除SCD1基因奶山羊核供体提供了生物材料.
