在1例Usher综合征家系中定位MYO7A基因的两个新突变位点
2019-03-21刘梦婷张天虹张硕影云中燕孟琪
刘梦婷 张天虹 张硕影 云中燕 孟琪
Usher综合征是一组以视网膜色素变性及不同程度的听力障碍为主要特征的常染色体隐性遗传性疾病[1],一些患者还伴有前庭功能障碍的表现。流行病学调查显示,不同人群中,Usher综合征的发病率大约在3.3~16.6/100000[2~4]。根据症状的严重程度将Usher综合征分为三型[5]。I型患者症状最重,主要表现为先天性重度到极重度感音神经性聋,前庭功能障碍,视网膜色素变性发生较早,一般在10岁左右发生。II型患者表现为先天性中度到重度感音神经性聋,前庭功能正常,视网膜色素变性发生较I型晚,通常在青春期发生[6]。III型通常表现为语后聋,逐渐加重的听力损失及前庭功能障碍,视网膜色素变性发生时间较晚,通常在20岁左右[7]。
Usher综合征遗传异质性较高,目前已定位14个相关基因组区间,发现致病基因10个。其中I型相关位点有9个,分别为USH1B-K。明确致病基因6个,分别为MYO7A、USH1C、CDH23、PCDH15、SANS和CIB2,其中MYO7A基因突变在Usher综合征I型中最为常见,约占53.2%[8]。本研究通过全外显子捕获测序技术结合软件分析和Sanger验证,在一个Usher综合征家系中发现了MYO7A基因复合杂合突变c.3412C>T(p.Q1138*)、c.4152G>C (p.K1384N),这两个突变位点在既往的文献中均未见报道,本研究拓展了MYO7A基因的突变谱。
1 对象与方法
1.1 研究对象
先证者为10岁女童,新生儿听力筛查初筛及复筛均未通过,3个月时于哈尔滨儿童医院行声导抗检查,结果为A型曲线。行听性脑干反应(ABR)检测,各给声强度V波均未引出。行多频稳态(ASSR)检查,双耳平均听阈均为100 dB HL,最终诊断为极重度感音神经性聋。患儿于2岁时行左侧人工耳蜗植入术。5岁时出现夜盲并逐渐加重,视野缩小,视力下降。7岁时于我院眼科就诊,行视力、眼底视野视网膜电图等相关检查。裸眼视力左眼0.3,右眼0.3。眼底检查显示视乳头变黄,视网膜血管狭窄,骨针样色素细胞沉积和斑点。视野检查显示环形缺损。视网膜电图显示各种闪光强度检查均为熄灭型。诊断为视网膜色素变性。先证者伴有前庭功能减退的证据,如20个月才学会走路,较正常儿童晚。根据先证者典型的临床表现,初步诊断为Usher综合征I型。对患儿一级亲属进行详细的体格检查,其父母无相似临床表现。(图1)
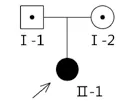
图1 家系谱图
1.2 研究方法
本研究遵循赫尔辛基宣言,经哈尔滨医科大学附属第一医院伦理委员会批准,在获得患者及其家属知情同意后,采集先证者及其父母静脉血3 mL(EDTA 抗凝血) 并进行致病基因突变分析。首先,对先证者进行基因全外显子及剪切位点测序,方法如下:(1)基因组DNA 提取:使用血液基因组DNA提取试剂盒,操作按照试剂盒说明书进行。(2)靶向捕获二代测序:针对基因组外显子区域采用安捷伦V6基因组外显子区域定制捕获探针进行目标基因全外显子捕获测序:①文库构建:首先使用非接触式超声波细胞粉碎机打断基因组DNA使其片段化至约150~200 bp,然后使用Kapa末端修复酶进行末端修复,将修复后的DNA使用AMPure磁珠进行纯化,并使用kapaA-tailing试剂在DNA 3′端添加A碱基,再使用KAPA DNA Ligase将Adapter连接到DNA片段上,最后进行文库模板量平衡的线性扩增,完成文库制备;②杂交捕获:真空浓缩DNA文库样本进行杂交捕获,PCR产物采用Ampure磁珠进行纯化,并进行Qubit定量;③测序:hiseq 2500平台标准化上机测序;④测序数据质量评估:测序原始数据经Illumina测序操控软件(sequence control software,SCS)评估合格;⑤数据分析:去除接头及低质量数据;获得clean data、基本数据;基因组分析工具包(the genome analysis toolkit,GATK)分析(variation calling);SNV和Indel突变位点汇总;比对正常人群及已知致病人群数据:关联人基因突变数据库(human gene mutation database,HGMD)/千人基因组/人类基因组变异整合数据库(genome aggregation database,gnomAD )/本地人群频率数据库;已知疾病的基因突变点汇总(千人基因组频率<5%且本地人群频率<2%);使用SIFT软件利用基于同源比对、蛋白结构保守性等的算法,预测筛选出变异对蛋白结构和功能的影响,对剪切位点附近的突变,做剪接危害性预测;参照ACMG指南,筛选与临床表型相关Pathogenic/Likely Pathogenic/Uncertain Significance的单核苷酸多态性(single nucleotide polymorphism,SNP)/Indel/exon基因拷贝数变异(copy number variations,CNV)。(3)一代测序(Sanger法)验证:将二代测序得到的高度可疑变异位点扩展至家系其他成员及100名正常对照进行验证,根据突变位点周围序列设计引物,采用聚合酶链式反应(PCR)进行扩增,PCR扩增产物用ABI 3730XL测序仪测序,基因序列分析采用DNASTAR软件进行序列分析和比对。
2 结果
2.1 家系表型特征
该家系共2代3人,仅先证者发病,系谱分析符合常染色体隐性遗传特征(见图1)。I-1、I-2否认近亲婚配,均无Usher综合征临床表现。先证者出生3个月诊断为极重度感音神经性听力损失。
2.2 基因检测结果
对先证者II-1行全外显子及剪切位点测序分析,结果显示MYO7A基因存在两个可疑突变c.3412C>T(p.Q1138*)、c.4152G>C (p.K1384N),经Sanger测序验证显示这两个突变分别来自先证者的父母(表1,图2)。

表1 家系的基因型结果
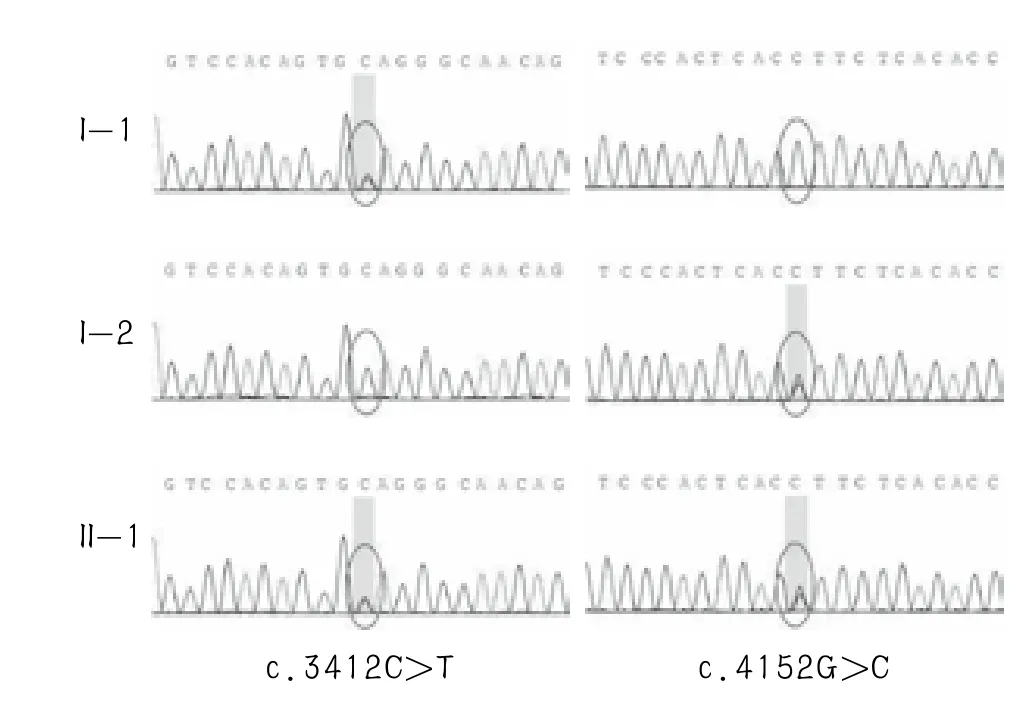
图2 家系Sanger测序结果
3 讨论
Usher综合征具有高度遗传异质性,已明确I型致病基因共有6个,其中MYO7A基因突变在I型中最常见。MYO7A基因突变与常染色体显性耳聋11型(AD)、常染色体隐性耳聋2型(AR)、Usher综合征IB型(AR)相关[9]。该基因由49个外显子组成,编码2215个氨基酸的myosinⅦA肌球蛋白,主要在内耳、视网膜、肾脏表达[10~11]。迄今为止,共发现导致Usher综合征I型的MYO7A突变位点505个,突变方式多样,包括错义突变、无义突变、移码突变、插入突变及剪切位点突变[12]。MYO7A基因无热点突变,因此发现该基因新的致病位点对于诊断Usher综合征I型至关重要。
笔者通过靶向捕获二代测序技术对疑似Usher综合征I型的一个中国小家系先证者进行了全基因外显子测序和数据分析比对后锁定MYO7A基因的两个突变分别为c.3412C>T(p.Q1138*)、c.4152G>C(p.K1384N)。经过一代测序验证两个突变分别来自于先证者父母,而I-1和I-2无相似临床表现,符合常染色体隐性遗传模式。其中c.3412C>T突变,导致蛋白质翻译提前中止p.Q1138*。HGMDpro数据库未见报道。根据ACMG指南,此变异类型评级为Pathogenic(致病性突变),该突变位于第21号外显子,为无义突变,导致蛋白翻译终止,对蛋白功能的影响较大,且有较低的人群携带率[13]。另一个突变点c.4152G>C,位于第31号外显子,属于错义突变,导致氨基酸改变p.K1384N。HGMDpro数据库未见报道。根据ACMG指南,其突变类型评级为Likely Pathogenic(可能致病性突变),笔者又采用多种预测软件对该位点致病性进一步预测,SIFT(http://sift.jcvi.org)预测该突变点为D:Deleterious(有害突变),PolyPhen-2(http://genetics.bwh.harvard.edu/pph2)预测结果为P:Possibly damaging(可能有害突变),MutationTaster(http://www.mutationtaster.org)预测结果为D:Disease_causing(致病性突变),进一步用限制性片段长度多态性分析方法对100个正常人进行分析,均未检测到这两个突变,再结合先证者的表型及基因型共分离分析,c.3412C>T(p.Q1138*)、c.4152G>C(p.K1384N)为该家系的致病突变。
近年来,随着高效安全的转染载体的不断发现,在动物模型中,针对Usher综合征的基因治疗成为可能。Pan[14]等将野生型的USH1C基因转染到带有Ush1c c.216G>A突变的USH1C小鼠模型的内耳中,惊喜的发现被转染小鼠的听觉和平衡行为的恢复几乎和野生型小鼠接近。Kevin Isgrig[15]等将野生型whirlin cDNA转染到带有whirler基因突变的Usher综合征小鼠中,发现在4个月后小鼠的听力和平衡能力都有明显的提高。动物模型基因治疗的成功也为人类Usher综合征的治疗带来了希望,但基因治疗依赖准确的临床诊断和分子学诊断。
综上所述,笔者在一个临床诊断为Usher综合征IB型的中国家系中发现MYO7A的两个新突变,c.3412C>T(p.Q1138*)、c.4152G>C (p.K1384N),根据ACMG指南和软件分析预测显示这两个突变为该家系的致病突变,本研究拓展了Usher综合征的突变谱,并对基因治疗提供了靶点,但对于它的致病机理还需要进一步研究。
