脂肪组织DNA甲基化与糖尿病和肥胖的发生发展
2019-02-28黄鑫陈永强徐国良彭淑红
黄鑫,陈永强,徐国良,彭淑红
脂肪组织DNA甲基化与糖尿病和肥胖的发生发展
黄鑫,陈永强,徐国良,彭淑红
江西中医药大学,中医基础理论分化发展研究中心,南昌 330004
糖尿病和肥胖是经常一起出现的复杂性代谢疾病,均受遗传因素和环境因素双重影响。越来越多的研究表明,DNA甲基化的变化会导致糖尿病和肥胖的发生。脂肪组织是糖尿病和肥胖主要的受累组织,同时作为内分泌器官参与多种病理生理过程,调节机体代谢平衡。脂肪组织相关基因的DNA甲基化变化会影响脂肪组织功能,从而与糖尿病和肥胖的发生密切相关。本文主要阐述糖尿病患者和肥胖患者不同类型脂肪组织DNA甲基化变化,以期深入了解糖尿病和肥胖发生发展的可能机制和探寻可能的防治途径。
糖尿病;脂肪组织;DNA甲基化
糖尿病和肥胖作为复杂性代谢疾病,发病率日趋增加。2017年,国际糖尿病联盟(international diabetes federation, IDF)公布的数据显示,目前全球有4.25亿糖尿病患者,预计到2045年,将会有近7亿糖尿病患者,其中95%以上是2型糖尿病(type 2 diabetes, T2D)。肥胖是糖尿病发生发展的主要危险因素,流行病学调查显示60%~90%的糖尿病患者为超重或肥胖。除遗传背景影响个体对糖尿病和肥胖患病的倾向外,饮食习惯、生活方式等也与其发病密切相关[1]。这些外界因素通过改变机体表观遗传学修饰(DNA甲基化、组蛋白修饰以及microRNA等),影响相关基因表达,从而影响机体相关功能。近年来,糖尿病和肥胖的表观遗传学机制逐渐成为研究热点。作为表观遗传学的一个重要研究内容,DNA甲基化是指在DNA甲基转移酶催化下,以S-腺苷甲硫氨酸作为甲基供体使CpG岛上胞嘧啶5′端甲基化。近年来,尽管国内外已有DNA甲基化与糖尿病相关研究进展的报道[2,3],但是较少系统阐述糖尿病、肥胖与脂肪组织DNA甲基化的关系。脂肪组织是机体重要储存能量和分泌激素的器官,其分布不同和功能紊乱会直接影响T2D以及其他代谢性综合征的发生,在体重指数(body mass index,BMI)水平相同时,上半身脂肪较多的个体患T2D风险明显增加[4,5],内脏脂肪组织(visceral adipose tissue, VAT)脂肪酸水平较浅层腹部皮下脂肪高,可以导致胰岛素抵抗增加[6]。而股臀皮下脂肪沉积相对于腹部内脏脂肪沉积,能降低T2D风险性[7]。因此探讨糖尿病和肥胖受累者不同脂肪组织DNA甲基化的变化,有助于深入了解糖尿病和肥胖的发生发展机制和探寻可能的防治途径。本文从不同类型脂肪组织入手,分别阐述糖尿病患者和肥胖患者在各类型脂肪组织中DNA甲基化的变化,并对存在的问题进行思考,旨在为糖尿病和肥胖的预防治疗寻找新途径。
1 脂肪组织的分类
哺乳动物脂肪组织根据形态、功能以及发育起源的不同可分为白色脂肪组织(white adipose tissue, WAT)和棕色脂肪组织(brown adipose tissue, BAT)。WAT呈黄色,在某些哺乳动物体内呈白色(如小鼠)。主要分布在皮下组织、网膜和肠系膜等处,是体内最大的能源库,参与脂肪代谢。根据分布差异又将WAT分为皮下脂肪组织(subcutaneous adipose tissue, SAT)和VAT。SAT是指分布于全身皮肤下层的脂肪组织,VAT主要指位于内脏周围的脂肪组织。BAT呈棕色,主要分布在新生儿肩胛间、腋窝和颈后部,成人BAT主要集中在锁骨上区、颈部、腋窝等处。BAT在代谢性疾病方面有着重要意义。另外,在某些刺激下(如冷暴露、β3肾上腺能激动剂等),白色脂肪可以向棕色样脂肪转化,这种棕色样脂肪被称为米色脂肪(beige fat)[8]。
2 糖尿病患者和肥胖患者SAT中DNA甲基化变化
SAT不仅是最大的脂肪储存组织,也是脂类储存最安全的位置,脂类物质在此储存对人体的危害最小。然而,其储存能力依赖于脂肪细胞肥大能力以及募集可以储存多余脂肪的新细胞的能力。当超过其储存能力时,脂肪就只能储存在皮下以外,如肝脏、骨骼肌和胰岛等,即所谓的脂肪异位沉积。脂肪的异位沉积是导致肥胖相关的胰岛素抵抗和炎症的重要因素。因此SAT储存脂肪的能力与肥胖以及糖尿病等代谢性疾病密切相关。在糖尿病或肥胖患者SAT中常常出现某些基因甲基化的异常,可能预示着脂肪组织功能的变化。
2.1 糖尿病患者SAT中DNA甲基化变化
在糖尿病患者SAT中,与胰岛素抵抗、脂类运输和脂肪生成、细胞增殖以及细胞分化相关的基因会发生DNA甲基化的变化(表1)。
2.1.1 糖尿病患者SAT中胰岛素抵抗相关基因甲基化变化
正常脂肪组织对于胰岛素高度敏感,胰岛素促进脂肪沉积(通过促进脂肪酸的吸收、重酯化和从头脂肪生成)并且抑制甘油三酯分解。而胰岛素抵抗的脂肪组织即使在高胰岛素水平下,也不能抑制甘油三酯分解,机体呈现葡萄糖不耐受性以及血浆游离脂肪酸水平的增加,而血浆游离脂肪酸增加会损伤肌肉胰岛素信号,促进肝脏糖异生,从而损伤葡萄糖刺激的胰岛素应答[9],导致系统胰岛素抵抗。
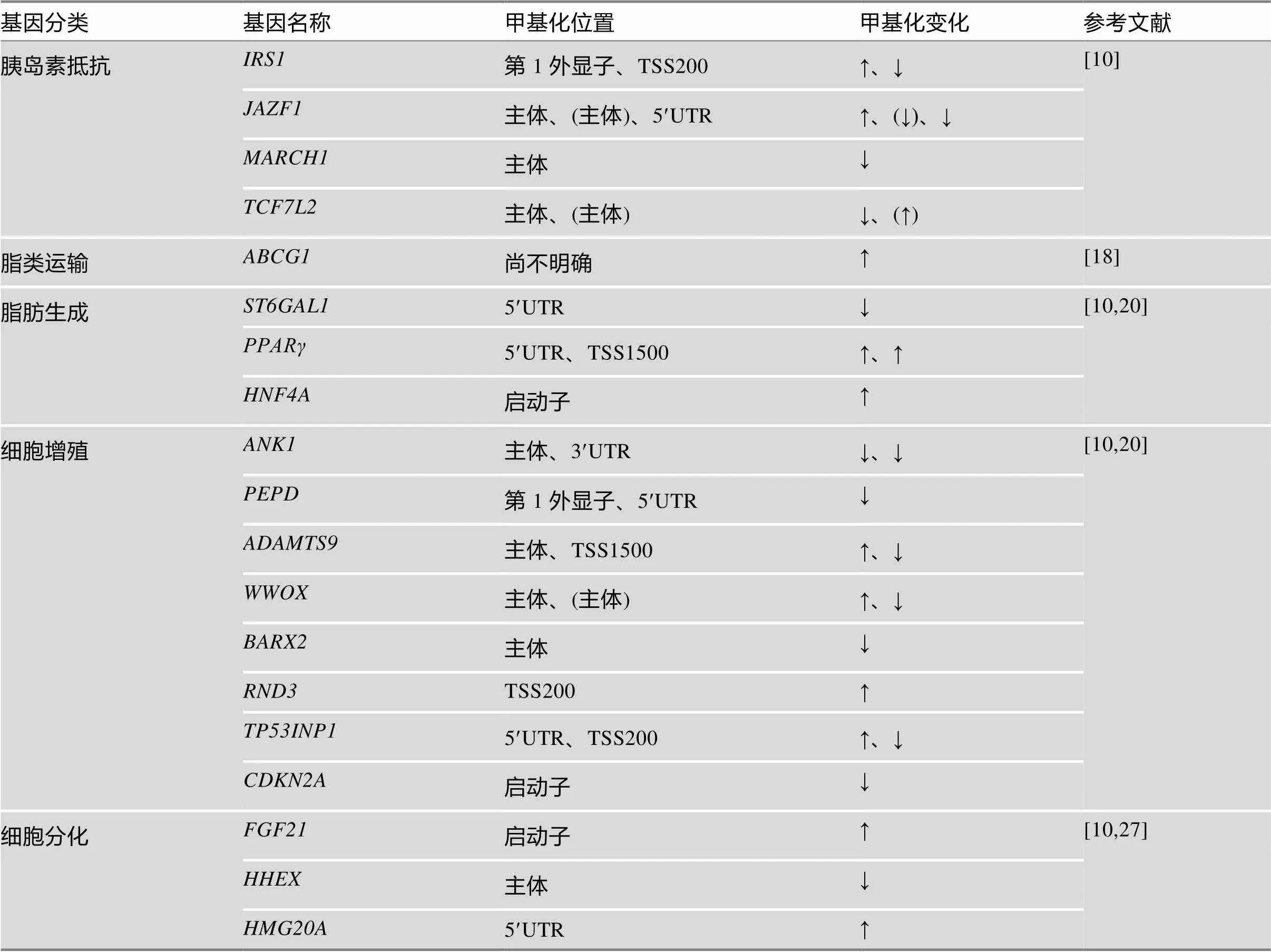
表1 糖尿病患者SAT中DNA甲基化变化
某些基因主体(body)区域有不同CpG位点,发生不一样的DNA甲基化变化,因此表中以“主体”以及“(主体)”标示作为区别。
尽管针对糖尿病SAT胰岛素抵抗基因甲基化的报道不多,但有少数研究组从全基因组甲基化的角度分析了糖尿病群体和非糖尿病群体SAT DNA甲基化的差异,发现与胰岛素抵抗相关的部分基因出现甲基化异常。
Nilsson等[10]分析了不同遗传背景下糖尿病群体和非糖尿病群体的SAT全基因组DNA甲基化,发现两个群体有差异DNA甲基化CpG位点,经KEGG分析,差异DNA甲基化的7046个基因,其中与胰岛素抵抗相关基因有、、和等。
IRS1在胰岛素信号中发挥重要作用,作为一种重要蛋白与磷酸化胰岛素受体结合。IRS1缺失会导致胰岛素抵抗[11]。敲除小鼠模型表明,该基因在脂肪分化中也发挥重要作用[12]。在糖尿病患者SAT脂肪组织中,基因第1外显子上5个CpG位点均呈现了高甲基化,只有在转录起始位点(transcription start site, TSS)200处的一个CpG位点表现出低甲基化[10]。而且,同时发现在糖尿病患者脂肪组织中表达水平显著降低[10]。另外,Orozco等[13]也报道,代谢综合征男性糖尿病患者第1外显子的CpG位点(cg17848496、cg00727310和cg14283647)均有显著的高甲基化。一般来说,DNA启动子区域甲基化表现出与表达呈负相关,而发生在基因主体(body)和3′非翻译区(3′ untranslated region, 3′UTR)的DNA甲基化与表达呈正相关[14]。而糖尿病患者人群SAT中第1外显子区域DNA甲基化,降低了该基因表达。
与糖异生、胰岛素抵抗、脂代谢和炎症均有关。该基因过表达可以增加胰岛素敏感性[15]。然而在糖尿病患者SAT组织中的基因主体区域CpG位点(cg06337342和cg04493169)甲基化水平增加,而在另一个基因主体区域CpG位点(cg14491535)和5′UTR 区域CpG位点(cg23620719)甲基化水平降低。究竟哪些CpG位点甲基化与该基因的表达密切相关仍不清楚。
是一种E3泛素化连接酶,该基因功能缺失会增强、其过表达会损伤小鼠肝脏胰岛素敏感性。会泛素化胰岛素受体(insulin receptor, INSR)而降低细胞表面INSR水平[16]。然而糖尿病SAT基因主体区域CpG位点的DNA甲基化水平降低。该位点DNA甲基化在脂肪组织中与基因表达水平的关系仍需进一步明确。
T2D患者SAT中短mRNA可变剪接体表达与禁食游离脂肪酸水平呈正相关,表明其影响了脂肪组织胰岛素作用[17]。糖尿病SAT在3个位于基因主体区域的CpG位点(cg24788483、cg03683087和cg05923857)甲基化水平降低,而在另一个基因主体CpG位点(cg03339956)甲基化水平升高。基因主体区域DNA甲基化除与基因表达有关,还可能与mRNA的剪接有关[14]。因此,糖尿病患者主体区域的甲基化位点增加或降低,预示其功能变化,然而究竟具体哪些甲基化位点如何影响到该基因功能,仍需进一步阐明。
2.1.2 糖尿病患者SAT中脂类运输和脂肪生成相关基因甲基化变化
在T2D患者SAT中的(cg06500161) DNA甲基化水平也有显著增加[18]。而ABCG1促进脂类(胆固醇、磷脂、鞘磷脂和氧化型胆固醇)运输,在维持脂类平衡中发挥重要作用[19]。SAT中高甲基化预示着脂肪组织脂类运输能力降低。
在T2D患者SAT中与脂肪生成有关的、和甲基化水平也有显著变化。在5′UTR区域CpG位点甲基化水平显著降低,5′UTR和TSS1500区域CpG位点甲基化水平显著增加,而启动子甲基化水平也显著增加[10,20]。在肥胖小鼠VAT显著下调,ST6GAL1对脂肪生成过程具有抑制作用[21]。其5′UTR区域甲基化水平降低,预示其表达水平增加,因此可能抑制SAT脂肪生成。而PPARγ有助于刺激脂肪细胞脂肪酸吸收和脂肪形成,敲除小鼠即使饲喂高脂饲料(high fat diet,HFD)也不能生成脂肪组织[22]。糖尿病SAT中高甲基化意味着SAT中摄取脂肪酸能力以及由前脂细胞向脂肪细胞分化能力下降。然而,在糖尿病小鼠(/)SAT中甲基化状态有不同报道,Fujiki等[23]发现糖尿病小鼠SAT的甲基化(-437 bp和-247 bp)降低,伴随基因mRNA水平的升高;而饮食诱导肥胖小鼠SAT中甲基化与对照小鼠没有显著变化。可能不同的种属以及不同内在和外在环境对PPARγ影响不同。HNF4A通过与丙酮酸羧化酶(pyruvate carboxylase, PC)基因启动子相关元件结合,从而调节该基因表达。如果抑制表达,会下调PC 60%mRNA和50%蛋白水平[24]。PC不仅在糖异生活跃组织中高表达,在白色脂肪组织中也有高水平表达[25],这可能和该酶参与脂生成有关,其产物草酰乙酸可以为脂肪酸合成提供重要的酰基基团和NADPH。因此,SAT中发生高甲基化,提示HNF4A减少,从而PC表达降低,因此降低SAT脂生成过程。
、和甲基化在糖尿病患者SAT中的变化提示了糖尿病SAT脂肪生成能力降低。
2.1.3 糖尿病患者SAT中细胞增殖相关基因甲基化变化
在T2D患者SAT中,与促进细胞增殖作用相关的和基因主体区域CpG位点甲基化水平显著降低;提示该基因在T2D SAT中转录水平受到抑制,因此可能降低了SAT脂肪细胞增殖作用。而与抑制细胞增殖作用相关的基因和在不同位点可能表现出不同甲基化变化[10,20]。在基因主体区域CpG位点甲基化显著增加,但TSS1500区域CpG位点甲基化显著降低;基因在启动子区域甲基化降低;有2个基因主体区域CpG位点甲基化降低,1个CpG位点升高;基因主体区域CpG位点甲基化降低;TSS200区域甲基化增加;5'UTR区域甲基化增加,TSS200区域甲基化降低。基因主体区域CpG甲基化增加以及启动子DNA甲基化降低,也提示对应基因转录活性增加,因此可能具有抑制细胞增殖的作用。
总之,糖尿病SAT中、和等基因甲基化的变化,降低了脂肪组织细胞的增殖作用。
2.1.4 糖尿病患者SAT中细胞分化相关基因甲基化变化
FGF21在脂肪细胞中有助于葡萄糖吸收和脂肪细胞分化,并且能刺激转录活性增强[26]。并且是DNA甲基转移酶DNMT3A的靶基因,体外研究也发现,在T2D患者SAT的脂肪细胞中,启动子区域DNA甲基化与对照比较有显著增加[27],同时其表达量显著降低。因此启动子DNA甲基化可能降低脂肪细胞分化能力。
另外,与细胞分化有关的和在糖尿患者群中也有不同甲基化变化,其中基因主体区域DNA甲基化水平升高,而5′UTR甲基化水平增加[10],都提示其基因转录水平降低,从而降低脂肪细胞分化能力。
总之,糖尿病患者SAT中与胰岛素抵抗、脂肪运输、脂肪生成、细胞增殖和分化有关基因甲基化的变化,提示糖尿病SAT脂肪细胞胰岛素抵抗增加、细胞增殖能力以及脂肪生成和分化能力降低。
正是基于DNA甲基化与T2D的密切关系,Orozco等[13]建立了基于SAT中DNA甲基化的生物标记物来评估T2D危险性。结果证明脂肪组织DNA甲基化对于了解代谢综合征的分子效应是一个强大工具,能很好地评估T2D发展的危险性。
2.2 肥胖患者和动物模型SAT中DNA甲基化变化
在肥胖患者或动物SAT中,常常出现与脂肪分化、形成有关的基因甲基化变化(表2),从而影响SAT功能。
2.2.1 肥胖患者SAT中DNA甲基化变化
Pietiläinen等[28]分析了26对BMI不一致的同卵双生双胞胎SAT中DNA甲基化和相关基因表达,发现了胖和瘦的双胞胎中有17个与肥胖相关的基因存在差异甲基化水平。这些基因主要涉及脂代谢、炎症和细胞外基质(extracellular matrix, ECM)重塑,并且与脂代谢相关基因表达下调,而炎症和ECM相关基因表达上调。Keller等[29]研究也发现,肥胖个体SAT中有46个基因与非肥胖个体甲基化显著不同,高甲基化的前10个基因为、、、、、、、、和,低甲基化的前10个基因、、、、、、、、和。Crujeiras等[30]研究发现肥胖患者SAT的差异甲基化CpG位点甲基化水平比非肥胖患者低。Dahlman等[31]分析了肥胖后(post-obese)妇女和从未肥胖(never-obese)妇女SAT的DNA甲基化,发现所有分析位点的平均甲基化程度,肥胖后妇女比从未肥胖的妇女低。此外,差异甲基化区域(differentially methylated region, DMR)主要位于CpG岛和及周边,8504个差异甲基化位点(differentially methylated sites, DMS)定位于3717个特定基因,其中27%是与脂生成有关。而Cordero等[32]研究了27位肥胖妇女SAT中和的甲基化,他们研究发现,经8周低卡路里饮食后,体重显著减轻妇女SAT中的和甲基化显著降低。另外,体重超重人SAT脂肪组织中的启动子甲基化水平与血浆高密度脂蛋白胆固醇呈负相关[33]。总之,在肥胖患者SAT中相关基因DNA甲基化变化可能反映了脂代谢降低、炎症发生。
2.2.2 肥胖动物模型SAT中DNA甲基化的变化
Sonne等[34]用Med-IP方法研究了饮食诱导肥胖鼠模型以及/鼠模型腹股沟白色脂肪组织(inguinal white adipose tissue, iWAT)(SAT的一种类型)的甲基化,发现在这两种模型中,SAT均出现了DNA低甲基化。饮食诱导的脂肪组织DNA甲基化变化涉及了脂生成基因表达。在iWAT中鉴定了39个基因的甲基化伴随有mRNA水平的变化,其中和基因表达水平降低,这两个基因均涉及了脂肪代谢。主要编码了脂肪细胞与脂分解有关的酶,而编码了使用酮体的酶,它为脂肪酸和胆固醇提供乙酰CoA。另有研究发现高脂食物诱导肥胖小鼠SAT(iWAT)中,表达显著降低,伴随其在分化脂肪细胞中出现DNA甲基化;因为具有促进脂肪细胞分化的作用,因此高脂饮食诱导肥胖小鼠SAT脂肪分化作用降低[35]。除此之外,也有促进脂肪细胞分化作用[36],高脂饮食也能诱导甲基化:Claycombe等[37]研究了出生后饮食高脂饲料的大鼠,发现其SAT中第1号染色体的Igf2/H19位点ICR/H19 差异甲基化区域,位点1~4的DNA甲基化相对于正常饮食有显著增加。总之,肥胖动物模型SAT中、、以及甲基化变化伴随其表达量降低,表明其脂肪分解能力、脂类合成能力以及脂肪分化能力降低。
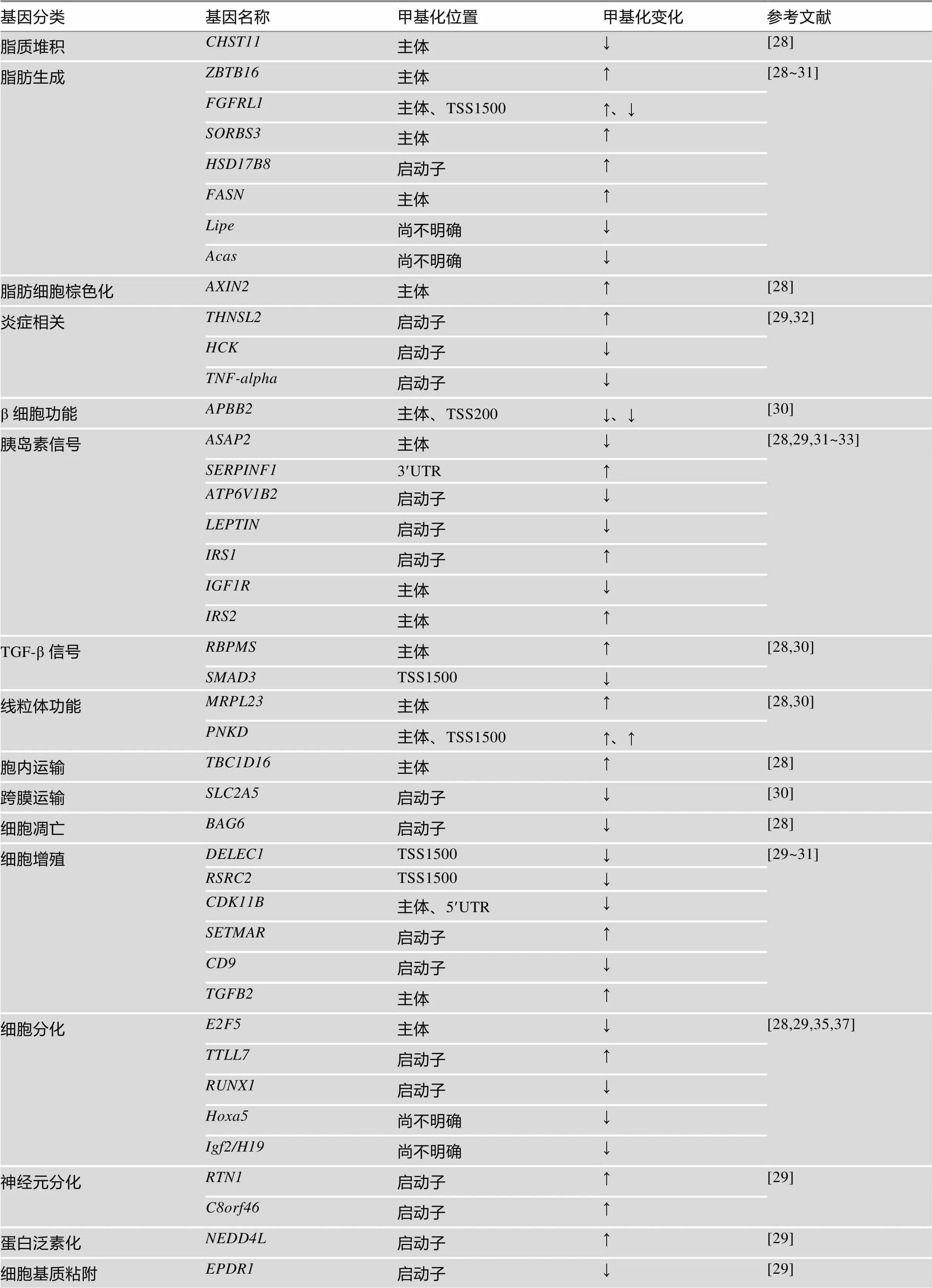
表2 肥胖患者及动物模型SAT中DNA甲基化的变化
3 糖尿病患者和肥胖患者VAT中DNA甲基化变化
相比于SAT,VAT有更多血管、神经,并包含更多数目的炎症、免疫细胞和更大比例的大脂肪细胞,而且脂肪细胞代谢更加活跃,但是其前脂细胞分化能力降低,对胰岛素表现出更强的抵抗[38]。内脏脂肪型肥胖往往与死亡率增加有很强的相关性[39]。在胰岛素抵抗的糖尿病前期患者中,VAT显著增加。并且VAT与胰岛素抵抗比SAT有更强的相关性[40]。因此糖尿病患者VAT变化可能反映于糖尿病整个发展过程中,而VAT中相关基因DNA甲基化与这种VAT组织与功能的变化可能有很大关系。
3.1 糖尿病患者VAT中DNA甲基化变化
RodrõÂguez-Rodero等[41]分析了经过胃肠绕道术的T2D和非T2D女性VAT DNA甲基化,发现T2D和非T2D在16个基因中有24个显著不同的CpGs。其中,基因高甲基化与T2D糖尿病显著相关。HOOK2主要参与中心体相关功能[42],但是其对于脂肪细胞相关的作用仍不清楚。Deng等[43]研究了孕期糖尿病妇女VAT中全基因组DNA甲基化和基因表达谱,发现、、、、、和有DNA甲基化变化,并且伴随基因表达的变化(表3)。
3.2 肥胖患者及肥胖动物模型VAT中DNA甲基化变化
有不少研究者从全基因组甲基化的角度研究肥胖患者及动物模型的VAT甲基化水平,相对于非肥胖个体其VAT甲基化差异基因主要集中在以下几个方面(表4)。
3.2.1 炎症基因甲基化变化
Keller等[29]分析了肥胖和非肥胖个体OVAT (omental visceral adipose tissue, OVAT)全基因组DNA甲基化,发现在肥胖个体中呈高甲基化。DUSP22是一个负调控因子,通过对STAT3去磷酸化调节IL-6/LIF/STAT3介导的信号通路,IL-6可以通过激活STAT3将与肥胖相关的慢性炎症与胰岛素抵抗联系在一起。过表达可以抑制IL-6诱导的STAT3磷酸化,而降低炎症发生[44,45],因此该基因甲基化的增加可能促进了炎症发生。然而有趣的是,同样作为炎症反向调节因子(敲除小鼠表现出慢性炎症的表型,一些炎症因子表达增加[46]),在肥胖个体中的甲基化水平降低[29]。并且在HFD诱导的肥胖小鼠附睾脂肪组织(epididymal white adipose tissue, eWAT)中表达增加[47]。和均为炎症的负调节因子,但是其在VAT表现出完全相反的甲基化变化,具体原因仍不清楚。
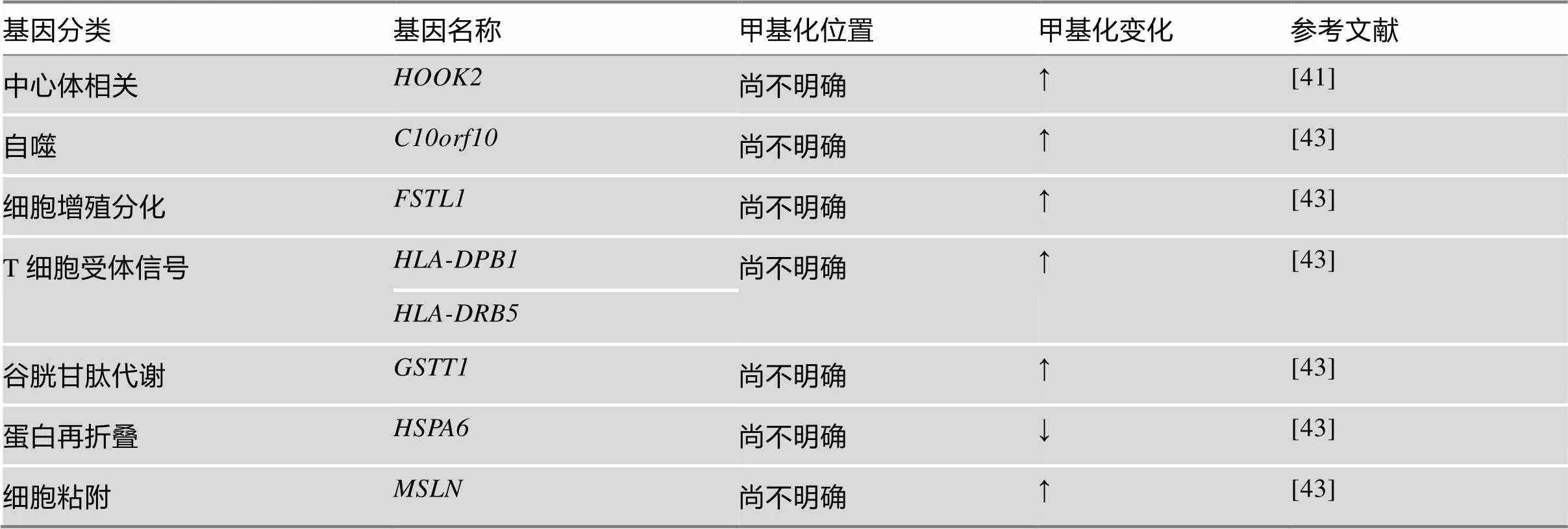
表3 糖尿病患者VAT中DNA甲基化的变化
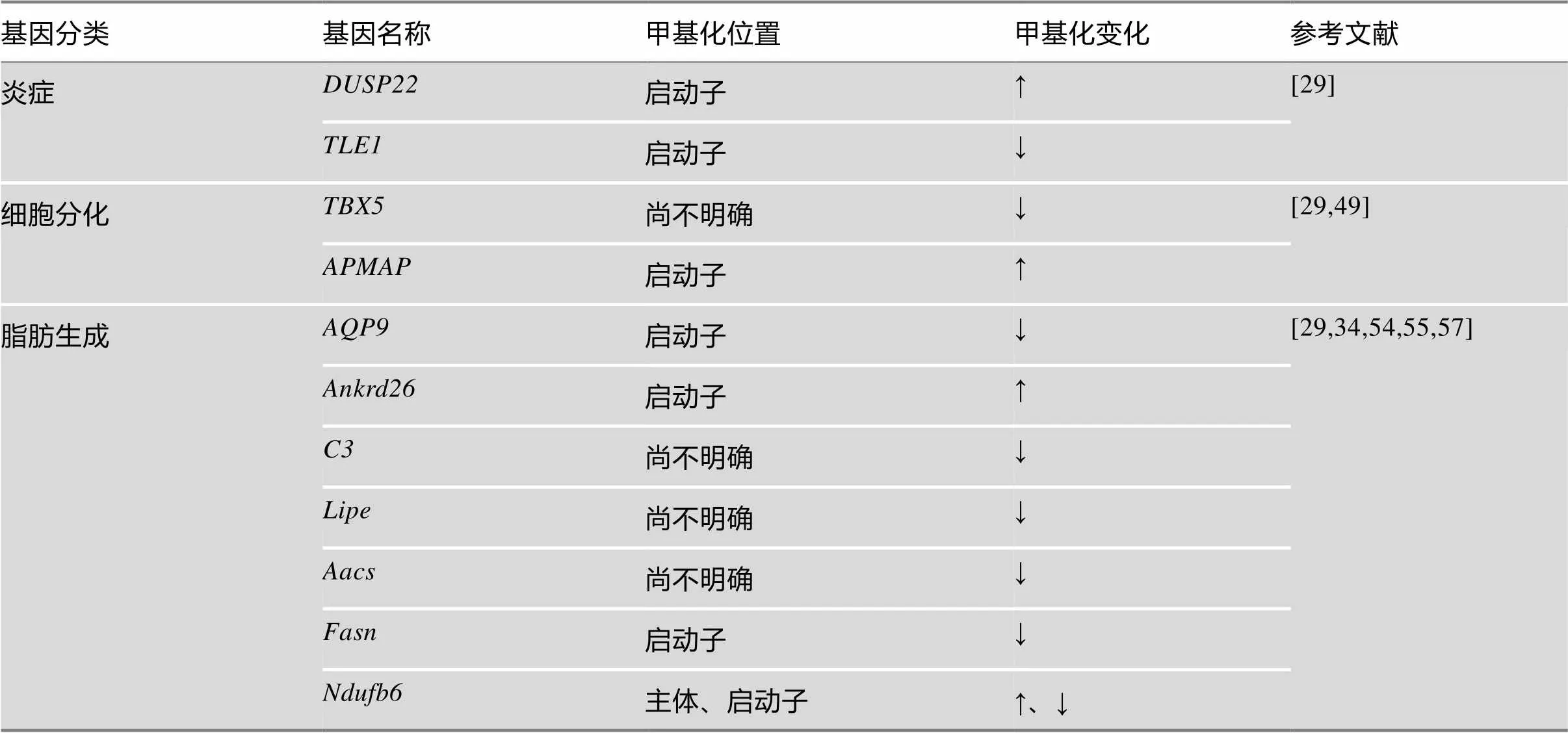
表4 肥胖患者及肥胖动物模型VAT中DNA甲基化的变化
3.2.2 细胞分化相关基因甲基化变化
与腹部脂肪组织的前脂细胞增殖和分化有关[48]。在IR抵抗人群中基因甲基化降低[49]。APMAP也是与脂肪细胞分化有关的蛋白,在3T3-L1细胞中沉默该基因表达会损伤前脂细胞向脂肪细胞分化[50]。在肥胖人群VAT中,该基因甲基化水平升高[29],预示其转录水平降低,从而可能损伤脂肪组织分化过程。
3.2.3 脂肪生成相关基因甲基化变化
基因在肥胖个体VAT呈现低甲基化[29]。AQP9作为甘油辅助转运体,主要在网膜和皮下脂肪表达从而促进甘油转出,在肝脏中辅助甘油转入用于糖异生;而胰岛素和瘦素能调节AQP9的活性,表达增加会促进脂肪生成[51,52]。肥胖个体VAT中,等基因低甲基化,预示着VAT在肥胖个体中脂肪生成增加。也是调节脂肪形成的重要因子之一,选择性降低该基因表达会促进3T3-L1细胞的脂肪形成[53]。HFD饮食会导致小鼠VAT中启动子在-436 bp和-431 bp的CpG位点出现特异性高甲基化,并损伤其表达[54]。该基因表达量降低预示其促进脂肪细胞脂肪的生成。另外,在过度肥胖人群VAT中,补体组分3(complement factor 3, C3) DNA甲基化降低,而该组分可以转化为促酰基化蛋白(acylation stimulating protein, ASP)从而刺激脂类储存[55]。
然而,也有一些报道显示与脂生成有关的基因在肥胖小鼠VAT中DNA甲基化水平降低或升高,但抑制了脂肪生成过程。如饮食诱导的肥胖模型鼠eWAT中,与脂生成有关的和甲基化水平降低,同时伴随有mRNA水平降低[34]。另外,编码产物是脂生成抑制因子[56],其在肥胖小鼠eWAT表达增加。这些基因的变化说明,在饮食诱导肥胖小鼠eWAT中,脂肪分解水平降低以及脂类合成降低。另一研究组以HFD饲喂大鼠,发现大鼠eWAT中和基因表达比对照组显著减低,且伴随基因-833/-829位点甲基化水平显著降低,基因在+143/+158位点甲基化显著增加,而在-7/+3/+14位点甲基化降低[57]。是脂肪酸合成酶基因,参与脂肪酸合成;而编码了呼吸链的重要组分,参与氧化磷酸化过程。和基因表达水平降低,提示HFD可能抑制了VAT脂肪酸合成以及物质的氧化分解。
4 棕色脂肪组织与DNA甲基化
BAT是通过适应性产热来调节体温的脂肪组织。BAT富含线粒体,而线粒体内膜上的解偶联蛋白(uncoupling protein 1, UCP1)能使氧化磷酸化解偶联,从而使原来ATP合成过程转换为以热的形式散失。然而,除了BAT,WAT经冷暴露或β肾上腺能受体激活(β-adrenergic receptor, β-AR)处理过后,脂肪细胞能表达UCP1,因此被称为米色脂肪[58]。
尽管鲜见报道糖尿病或肥胖患者BAT中DNA甲基化变化,但是由于BAT或米色脂肪组织在产热方面的能力,所以通过调节BAT或米色脂肪组织来改善肥胖或肥胖相关疾病如糖尿病引起人们的关注。
相对于WAT,BAT表达丰富的PRDM16 (co-regulator PR domain containing 16, PRDM16),该蛋白与其他转录因子PPARγ、PGC1α、C/EBPβ以及Zfp516调节BAT特异性基因如、(deionidase2,)以及(cell death-inducing DNA fragmentation factor alpha-like effector A,)的表达[59]。PRDM16是棕色和米色脂肪细胞中调节产热基因转录的重要因子,无论对于胚胎BAT发育以及成人BAT特异性基因的表达都是必要的[60]。PRDM16与典型的DNA转录因子如PPARγ和C/EBPβ形成转录复合物,特异性激活BAT选择性基因表达程序[61,62]。PRDM16转录起始位点含有丰富的CpG位点,可以受到DNA甲基化调控[63]。转录因子基因[64]、[65]、[66]以及BAT组织特异性表达基因[67]、[68]均可以受DNA甲基化调控。
因此通过调节BAT或米色脂肪组织特异性表达基因或者相关转录因子的DNA甲基化变化,从而可以促进这些脂肪组织产热,这也是许多研究者正在探寻的一种策略,用于治疗包括糖尿病在内的代谢性疾病。
5 结语与展望
糖尿病或肥胖患者在SAT和VAT中的DNA甲基化变化往往与脂肪生成、脂肪细胞分化以及炎症相关基因的变化有关(图1)。但目前有关糖尿病或肥胖患者BAT中DNA甲基化变化的报道较少。由于BAT利用脂肪的积极作用,人们将其作为有效治疗糖尿病或肥胖的一个组织靶点,正在探寻可能的治疗策略。
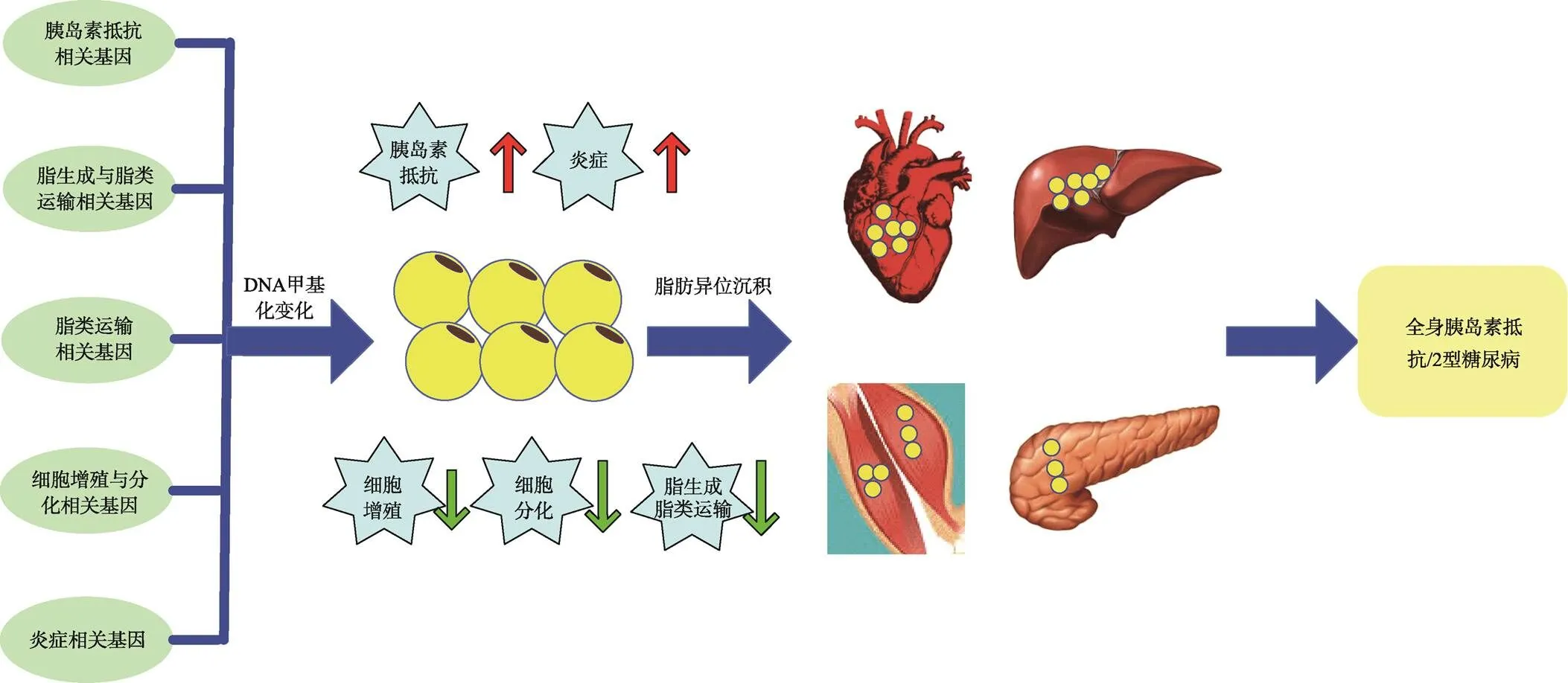
图1 脂肪组织相关基因DNA甲基化与胰岛素抵抗/2型糖尿病的关联示意图
尽管有很多临床和实验文章报道了糖尿病和肥胖脂肪组织DNA甲基化变化,但目前仍存在一些问题尚待深入研究:(1)脂肪组织中DNA甲基化频率与肥胖、糖尿病病变程度的确切关系是怎样的?(2)脂肪组织分布广泛,SAT、VAT和BAT在机体中所承担的角色各有不同,每个组织是否有特异表达基因,肥胖和糖尿病患者这些基因的甲基化情况是否代表着组织功能的变化?(3)DNA甲基化发生在基因的不同位置(启动子、增强子、转录区、基因间隔区等)对于基因表达调控不同,肥胖和糖尿病同一脂肪组织不同基因位置甲基化与基因表达关系图谱是怎样的?(4)脂肪组织包含多种细胞类型,而DNA甲基化有细胞特异性,究竟是脂肪细胞DNA甲基化,还是其他细胞甲基化?如何有效区分将更能反映组织功能变化情况?(5)脂肪组织的DNA甲基化与其它发生胰岛素抵抗的组织(如肝脏、骨骼肌等)的DNA甲基化是否有关?两者之间有无相互影响?
总之,脂肪组织不仅是合成及储存脂肪的仓库,更是参与多种病理生理过程的内分泌器官,对于机体代谢平衡的调节具有重要影响。脂肪组织DNA甲基化与肥胖、糖尿病发生发展密切相关。由于肥胖、糖尿病发病机制的复杂性、脂肪组织分布的广泛性、类型多样性以及DNA甲基化组织细胞特异性等决定了肥胖、糖尿病与脂肪组织DNA甲基化关系仍需要进一步研究。这些具体关系的研究将有助于糖尿病发病机制的阐明,也必将积极推动基于调节DNA甲基化的治疗肥胖和糖尿病药物的开发和筛选。
[1] Franks PW, Mccarthy MI. Exposing the exposures responsible for type 2 diabetes and obesity., 2016, 354(6308): 69–73.
[2] Mello VDFD, Pulkkinen L, Lalli M, Kolehmainen M, Pihlajamaki J, Uusitupa M. DNA methylation in obesity and type 2 diabetes., 2014, 46(3): 103–113.
[3] Tang LL, Liu Q, Bu SZ, Xu LT, Wang QW, Mai YF, Duan SW. The effect of environmental factors and DNA methylation on type 2 diabetes mellitus., 2013, 35(10): 1143–1152.汤琳琳, 刘琼, 步世忠, 徐雷艇,王钦文, 麦一峰, 段世伟. 2型糖尿病环境因素与DNA甲基化的研究进展. 遗传, 2013, 35(10): 1143–1152.
[4] Vague J. The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease., 1956, 4(1): 20– 34.
[5] Pi-Sunyer FX. The epidemiology of central fat distribution in relation to disease., 2004, 62(s2): S120–S126.
[6] Mårin P, Andersson B, Ottosson M, Olbe L, Chowdhury B, Kvist H, Holm G, Sjöström L, Björntorp P. The morphology and metabolism of intraabdominal adipose tissue in men., 1992, 41(11): 1242–1248.
[7] Tatsukawa Y, Misumi M, Kim YM, Yamada M, Ohishi W, Fujiwara S, Nakanishi S, Yoneda M. Body composition and development of diabetes: a 15-year follow-up study in a Japanese population., 2018, 72(3): 374–380.
[8] Seale P, Kajimura S, Spiegelman BM. Transcriptional control of brown adipocyte development and physiological function—of mice and men., 2009, 23(7): 788–797.
[9] Gastaldelli A, Gaggini M, Defronzo RA. Role of adipose tissue insulin resistance in the natural history of type 2 diabetes: results from the san antonio metabolism study., 2017, 66(4): 815–822.
[10] Nilsson E, Jansson PA, Perfilyev A, Volkov P, Pedersen M, Svensson MK, Poulsen P, Ribel-Madsen R, Pedersen NL, Almgren P, Fadista J, Rönn T, Pedersen KB, Scheele C, Vaag A, Ling C. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes., 2014, 63(9): 2962–2976.
[11] Guo S. Insulin signaling, resistance, and metabolic syndrome: insights from mouse models into disease mechanisms., 2014, 220(2): T1–T23.
[12] Miki H, Yamauchi T, Suzuki R, Komeda K, Tsuchida A, Kubota N, Terauchi Y, Kamon J, Kaburagi Y, Matsui J, Akanuma Y, Nagai R, Kimura S, Tobe K, Kadowaki T. Essential role of insulin receptor substrate 1 (IRS-1) and IRS-2 in adipocyte differentiation., 2001, 21(7): 2521–2532.
[13] Orozco LD, Farrell C, Hale C, Rubbi L, Rinaldi A, Civelek M, Pan C, Lam L, Montoya D, Edillor C, Seldin M, Boehnke M, Mohlke KL, Jacobsen S, Kuusisto J, Laakso M, Lusis AJ, Pellegrini M. Epigenome-wide association in adipose tissue from the METSIM cohort., 2018, 27(10): 1830–1846.
[14] Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond., 2012, 13(7): 484–492.
[15] Yuan L, Luo X, Zeng M, Zhang Y, Yang M, Zhang L, Liu R, Boden G, Liu H, Ma ZA, Li L, Yang G. Transcription factor TIP27 regulates glucose homeostasis and insulin sensitivity in a PI3-kinase/Akt-dependent manner in mice., 2015, 39(6): 949–958.
[16] Nagarajan A, Petersen MC, Nasiri AR, Butrico G, Fung A, Ruan HB, Kursawe R, Caprio S, Thibodeau J, Bourgeois- Daigneault MC, Sun L, Gao G, Bhanot S, Jurczak MJ, Green MR, Shulman GI, Wajapeyee N. MARCH1 regulates insulin sensitivity by controlling cell surface insulin receptor levels., 2016, 7: 12639.
[17] Kaminska D, Kuulasmaa T, Venesmaa S, Käkelä P, Vaittinen M, Pulkkinen L, Pääkkönen M, Gylling H, Laakso M, Pihlajamäki J. Adipose tissue TCF7L2 splicing is regulated by weight loss and associates with glucose and fatty acid metabolism., 2012, 61(11): 2807–2813.
[18] Dayeh T, Tuomi T, Almgren P, Perfilyev A, Jansson PA, de Mello VD, Pihlajamäki J, Vaag A, Groop L, Nilsson E, Ling C. DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk., 2016, 11(7): 482–488.
[19] Hardy LM, Frisdal E, Le Goff W. Critical role of the human ATP-binding cassette G1 transporter in cardiometabolic diseases., 2017, 18(9): 1892.
[20] Ribel-Madsen R, Fraga MF, Jacobsen S, Bork-Jensen J, Lara E, Calvanese V, Fernandez AF, Friedrichsen M, Vind BF, Højlund K, Beck-Nielsen H, Esteller M, Vaag A, Poulsen P. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes., 2012, 7(12): e51302.
[21] Kaburagi T, Kizuka Y, Kitazume S, Taniguchi N. The inhibitory role of α2,6-sialylation in adipogenesis., 2017, 292(6): 2278–2286.
[22] Jones JR, Barrick C, Kim KA, Lindner J, Blondeau B, Fujimoto Y, Shiota M, Kesterson RA, Kahn BB, Magnuson M A. Deletion of PPARγ in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance., 2005, 102(17): 6207–6212.
[23] Fujiki K, Kano F, Shiota K, Murata M. Expression of the peroxisome proliferator activated receptor γ gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes., 2009, 7: 38.
[24] Chavalit T, Rojvirat P, Muangsawat S, Jitrapakdee S. Hepatocyte nuclear factor 4αregulates the expression of the murine pyruvate carboxylase gene through the HNF4-specific binding motif in its proximal promoter., 2013, 1829(10): 987–999.
[25] Lynch CJ, Mccall KM, Billingsley ML, Bohlen LM, Hreniuk SP, Martin LF, Witters LA, Vannucci SJ. Pyruvate carboxylase in genetic obesity., 1992, 262(5): E608–E618.
[26] Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones., 2012, 148(3): 556–567.
[27] You D, Nilsson E, Tenen DE, Lyubetskaya A, Lo JC, Jiang R, Deng J, Dawes BA, Vaag A, Ling C, Rosen ED, Kang S. Dnmt3a is an epigenetic mediator of adipose insulin resistance., 2017, 6: e30766.
[28] Pietiläinen KH, Ismail K, Järvinen E, Heinonen S, Tummers M, Bollepalli S, Lyle R, Muniandy M, Moilanen E, Hakkarainen A, Lundbom J, Lundbom N, Rissanen A, Kaprio J, Ollikainen M. DNA methylation and gene expression patterns in adipose tissue differ significantly within young adult monozygotic BMI-discordant twin pairs., 2016, 40(4): 654–661.
[29] Keller M, Hopp L, Liu X, Wohland T, Rohde K, Cancello R, Klös M, Bacos K, Kern M, Eichelmann F, Dietrich A, Schön MR, Gärtner D, Lohmann T, Dreßler M, Stumvoll M, Kovacs P, Diblasio AM, Ling C, Binder H, Blüher M, Böttcher Y. Genome-wide DNA promoter methylation and transcriptome analysis in human adipose tissue unravels novel candidate genes for obesity., 2017, 6(1): 86–100.
[30] Crujeiras AB, Diaz-Lagares A, Sandoval J, Milagro FI, Navas-Carretero S, Carreira MC, Gomez A, Hervas D, Monteiro MP, Casanueva FF, Esteller M, Martinez JA. DNA methylation map in circulating leukocytes mirrors subcutaneous adipose tissue methylation pattern: a genome- wide analysis from non-obese and obese patients., 2017, 7: 41903.
[31] Dahlman I, Sinha I, Gao H, Brodin D, Thorell A, Rydén M, Andersson DP, Henriksson J, Perfilyev A, Ling C, Dahlman-Wright K, Arner P. The fat cell epigenetic signature in post-obese womenischaracterized by global hypomethylation and differential DNA methylation of adipogenesis genes., 2015, 39(6): 910–919.
[32] Cordero P, Campion J, Milagro FI, Goyenechea E, Steemburgo T, Javierre BM, Martinez JA. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet., 2011, 67(3): 463–470.
[33] Rohde K, Klös M, Hopp L, Liu X, Keller M, Stumvoll M, Dietrich A, Schön MR, Gärtner D, Lohmann T, Dreßler M, Kovacs P, Binder H, Blüher M, Böttcher Y. IRS1 DNA promoter methylation and expression in human adipose tissue are related to fat distribution and metabolic traits., 2017, 7(1): 12369.
[34] Sonne SB, Yadav R, Yin G, Dalgaard MD, Myrmel LS, Gupta R, Wang J, Madsen L, Kajimura S, Kristiansen K. Obesity is associated with depot-specific alterations in adipocyte DNA methylation and gene expression., 2017, 6(2): 124–133.
[35] Cao W, Xu Y, Luo D, Saeed M, Sun C. Hoxa5 promotes adipose differentiation via increasing DNA methylation level and inhibiting PKA/HSL signal pathway in mice., 2018, 45(3): 1023–1033.
[36] Kleiman A, Keats EC, Chan NG, Khan ZA. Elevated IGF2 prevents leptin induction and terminal adipocyte differentiation in hemangioma stem cells., 2013, 94(1): 126–136.
[37] Claycombe KJ, Uthus EO, Roemmich JN, Johnson LK, Johnson WT. Prenatal low-protein and postnatal high-fat diets induce rapid adipose tissue growth by inducing Igf2 expression in Sprague Dawley rat offspring., 2013, 143(10): 1533–1539.
[38] Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences., 2010, 11(1): 11–18.
[39] Pischon T, Boeing H, Hoffmann K, Bergmann M, Schulze MB, Overvad K, van der Schouw YT, Spencer E, Moons KGM, Tjønneland A, Halkjaer J, Jensen MK, Stegger J, Clavel-Chapelon F, Boutron-Ruault MC, Chajes V, Linseisen J, Kaaks R, Trichopoulou A, Trichopoulos D, Bamia C, Sieri S, Palli D, Tumino R, Vineis P, Panico S, Peeters PH, May AM, Bueno-De-Mesquita HB, van Duijnhoven FJ, Hallmans G, Weinehall L, Manjer J, Hedblad B, Lund E, Agudo A, Arriola L, Barricarte A, Navarro C, Martinez C, Quirós JR, Key T, Bingham S, Khaw KT, Boffetta P, Jenab M, Ferrari P, Riboli E. General and abdominal adiposity and risk of death in Europe., 2008, 359(20): 2105–2120.
[40] Liu L, Feng J, Zhang G, Yuan X, Li F, Yang T, Hao S, Huang D, Hsue C, Lou Q. Visceral adipose tissue is more strongly associated with insulin resistance than subcutaneous adipose tissue in Chinese subjects with pre-diabetes., 2018, 34(1): 123–129.
[41] Rodríguez-Rodero S, Menéndez-Torre E, Fernández-Bayón G, Morales-Sánchez P, Sanz L, Turienzo E, González JJ, Martinez-Faedo C, Suarez-Gutiérrez L, Ares J, Díaz-Naya L, Martin-Nieto A, Fernández-Morera JL, Fraga MF, Delgado-Álvarez E. Altered intragenic DNA methylation of HOOK2 gene in adipose tissue from individuals with obesity and type 2 diabetes., 2017, 12(12): e0189153.
[42] Pallesi-Pocachard E, Bazellieres E, Viallat-Lieutaud A, Delgrossi MH, Barthelemy-Requin M, Le Bivic A, Massey-Harroche D. Hook2, a microtubule-binding protein, interacts with Par6α and controls centrosome orientation during polarized cell migration., 2016, 6: 33259.
[43] Deng X, Yang Y, Sun H, Qi W, Duan Y, Qian Y. Analysis of whole genome-wide methylation and gene expression profiles in visceral omental adipose tissue of pregnancies with gestational diabetes mellitus., 2018, 81(7): 623–630.
[44] Sekine Y, Tsuji S, Ikeda O, Sato N, Aoki N, Aoyama K, Sugiyama K, Matsuda T. Regulation of STAT3-mediated signaling by LMW-DSP2., 2006, 25(42): 5801– 5806.
[45] Serrano-Marco L, Rodríguez-Calvo R, Kochairi IK, Palomer X, Michalik L, Wahli W, Vázquez-Carrera M. Activation of peroxisome proliferator-activated receptor-β/-δ (PPAR-β/-δ) ameliorates insulin signaling and reduces SOCS3 levels by inhibiting STAT3 in interleukin-6- stimulated adipocytes., 2011, 60(7): 1990–1999.
[46] Ramasamy S, Saez B, Mukhopadhyay S, Ding D, Ahmed AM, Chen X, Pucci F, Yamin R, Wang J, Pittet MJ, Kelleher CM, Scadden DT, Sweetser DA. Tle1 tumor suppressor negatively regulates inflammation in vivo and modulates NF-κB inflammatory pathway., 2016, 113(7): 1871–1876.
[47] Choi MS, Kim YJ, Kwon EY, Ryoo JY, Kim SR, Jung UJ. High-fat diet decreases energy expenditure and expression of genes controlling lipid metabolism, mitochondrial function and skeletal system development in the adipose tissue, along with increased expression of extracellular matrix remodelling- and inflammation-related genes., 2015, 113(6): 867–877.
[48] Pinnick KE, Nicholson G, Manolopoulos KN, Mcquáid SE, Valet P, Frayn KN, Denton N, Min JL, Zondervan KT, Fleckner J, Mccarthy MI, Holmes CC, Karpe F. Distinct developmental profile of lower-body adipose tissue defines resistance against obesity-associated metabolic complications., 2014, 63(11): 3785–3797.
[49] Crujeiras AB, Diaz-Lagares A, Moreno-Navarrete JM, Sandoval J, Hervas D, Gomez A, Ricart W, Casanueva FF, Esteller M, Fernandez-Real JM. Genome-wide DNA methylation pattern in visceral adipose tissue differentiates insulin-resistant from insulin-sensitive obese subjects., 2016, 178: 13–24.
[50] Bogner-Strauss JG, Prokesch A, Sanchez-Cabo F, Rieder D, Hackl H, Duszka K, Krogsdam A, Di Camillo B, Walenta E, Klatzer A, Lass A, Pinent M, Wong WC, Eisenhaber F, Trajanoski Z. Reconstruction of gene association network reveals a transmembrane protein required for adipogenesis and targeted by PPARγ., 2010, 67(23): 4049–4064.
[51] Ohgusu Y, Ohta KY, Ishii M, Katano T, Urano K, Watanabe J, Inoue K, Yuasa H. Functional characterization of human aquaporin 9 as a facilitative glycerol carrier., 2008, 23(4): 279–284.
[52] Rodríguez A, Catalán V, Gómez-Ambrosi J, Frühbeck G. Aquaglyceroporins serve as metabolic gateways in adiposity and insulin resistance control., 2011, 10(10): 1548–1556.
[53] Liu XF, Bera TK, Kahue C, Escobar T, Fei Z, Raciti GA, Pastan I. ANKRD26 and its interacting partners TRIO, GPS2, HMMR and DIPA regulate adipogenesis in 3T3-L1 cells., 2012, 7(5): e38130.
[54] Raciti GA, Spinelli R, Desiderio A, Longo M, Parrillo L, Nigro C, D'Esposito V, Mirra P, Fiory F, Pilone V, Forestieri P, Formisano P, Pastan I, Miele C, Beguinot F. Specific CpG hyper-methylation leads to Ankrd26 gene down-regulation in white adipose tissue of a mouse model of diet-induced obesity., 2017, 7: 43526.
[55] Castellano-Castillo D, Moreno-Indias I, Fernandez-Garcia JC, Clemente-Postigo M, Castro-Cabezas M, Tinahones FJ, Queipo-Ortuño MI, Cardona F. Complement factor C3 methylation and mRNA expression is associated to BMI and insulin resistance in obesity., 2018, 9(8): 410.
[56] Kang S, Akerblad P, Kiviranta R, Gupta RK, Kajimura S, Griffin MJ, Min J, Baron R, Rosen ED. Regulation of early adipose commitment by Zfp521., 2012, 10(11): e1001433.
[57] Lomba A, Martinez JA, Garcia-Diaz DF, Paternain L, Marti A, Campion J, Milagro FI. Weight gain induced by an isocaloric pair-fed high fat diet: a nutriepigenetic study on FASN and NDUFB6 gene promoters., 2010, 101(2–3): 273–278.
[58] Ishibashi J, Seale P. Beige can be slimming., 2010, 328(5982): 1113–1114.
[59] Sambeat A, Gulyaeva O, Dempersmier J, Sul HS. Epigenetic regulation of the thermogenic adipose program., 2017, 28(1): 19–31.
[60] Harms MJ, Ishibashi J, Wang W, Lim HW, Goyama S, Sato T, Kurokawa M, Won KJ, Seale P. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice., 2014, 19(4): 593–604.
[61] Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, Spiegelman BM. Initiation of myoblast to brown fat switch by a PRDM16-C/EBPβ transcriptional complex., 2009, 460(7259): 1154–1158.
[62] Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scimè A, Devarakonda S, Conroe HM, Erdjument- Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM. PRDM16 controls a brown fat/skeletal muscle switch., 2008, 454(7207): 961–967.
[63] Yang Q, Liang X, Sun X, Zhang L, Fu X, Rogers CJ, Berim A, Zhang S, Wang S, Wang B, Foretz M, Viollet B, Gang DR, Rodgers BD, Zhu MJ, Du M. Ampk/α- ketoglutarate axis dynamically mediates DNA demethylation in the prdm16 promoter and brown adipogenesis., 2016, 24(4): 542–554.
[64] Ma Y, Yang J, Wan Y, Peng Y, Ding S, Li Y, Xu B, Chen X, Xia W, Ke Y, Xu S. Low-level perfluorooctanoic acid enhances 3T3-L1 preadipocyte differentiation via altering peroxisome proliferator activated receptor gamma expression and its promoter DNA methylation., 2018, 38(3): 398–407.
[65] Clarke-Harris R, Wilkin TJ, Hosking J, Pinkney J, Jeffery AN, Metcalf BS, Godfrey KM, Voss LD, Lillycrop KA, Burdge GC. PGC1α promoter methylation in blood at 5-7 years predicts adiposity from 9 to 14 years (EarlyBird 50)., 2014, 63(7): 2528–2537.
[66] Rui W, Jin Z, Zhe G, Song H. The methylation of C/EBP β gene promoter and regulated by GATA-2 protein., 2013, 40(2): 797–801.
[67] Shore A, Karamitri A, Kemp P, Speakman JR, Lomax MA. Role of Ucp1 enhancer methylation and chromatin remodelling in the control of Ucp1 expression in murine adipose tissue., 2010, 53(6): 1164–1173.
[68] Huang YW, Luo J, Weng YI, Mutch DG, Goodfellow PJ, Miller DS, Huang TH. Promoter hypermethylation of CIDEA, HAAO and RXFP3 associated with microsatellite instability in endometrial carcinomas., 2010, 117(2): 239–247.
DNA methylation in adipose tissue and the development of diabetes and obesity
Xin Huang, Yongqiang Chen, Guoliang Xu, Shuhong Peng
Diabetes and obesity are complicated metabolic diseases which frequently occur together and are affected by environmental, hereditary and metabolic factors. Adipose tissue is involved in various physiological and pathological processes and plays an essential role as an endocrine organ which regulates the metabolic balance of the body. DNA methylation of some genes in adipose tissue may have an impact on its function. A growing body of evidence suggests that changes in DNA methylation may alter gene expression and lead to the development of diabetes and obesity in which adipose tissue function is imbalanced. This review discusses recent advances in alterations of DNA methylation in different types of adipose tissue in individuals with diabetes and obesity. This evidence may lead to a greater understanding of the pathogenesis of these diseases and lead to potential therapeutic interventions and management strategies for diabetes and obesity.
diabetes; adipose tissue; DNA methylation
2018-09-10;
2018-11-03
国家自然科学基金项目(编号:81503315,81560744)和国家留学基金委项目(编号:201608360175)资助 [Supported by the National Natural Science Foundation of China (Nos. 81503315, 81560744) and China Scholarship Council (No. 201608360175)]
黄鑫,硕士研究生,专业方向:中医药防治代谢性疾病。E-mail: 1103763638@qq.com
彭淑红,博士,副教授,研究方向:中医药防治代谢性疾病。E-mail: yaomoon@126.com
10.16288/j.yczz.18-206
2018/12/6 11:18:51
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20181206.1118.006.html
(责任编委: 陈雁)
