肺癌恶性胸腔积液中稀有肿瘤细胞的鉴定与单细胞测序分析
2019-02-28吴保军王卓董宇邓宇亮施奇惠
吴保军,王卓,董宇,邓宇亮,施奇惠
肺癌恶性胸腔积液中稀有肿瘤细胞的鉴定与单细胞测序分析
吴保军,王卓,董宇,邓宇亮,施奇惠
上海交通大学系统生物医学研究院,系统生物医学教育部重点实验室,上海 200240
肿瘤细胞的异质性是指肿瘤细胞在基因组或表型水平上具有不同特征。在外界环境压力或人为杀伤因素刺激下,肿瘤细胞具有不同的响应方式,从而导致它们在细胞增殖、侵袭转移及耐药能力等方面产生差别,尤其是一部分具有转移能力的肿瘤细胞能脱离原位组织并在远处器官形成转移灶。因此,肿瘤细胞的异质性为其发生转移和耐药提供了可能。传统的肿瘤异质性研究主要基于不同位置原位组织样本中的群体细胞,缺乏单细胞层面的解析,尤其是对具有转移能力的肿瘤细胞的异质性研究。本研究建立了基于肺癌恶性胸腔积液中转移性肿瘤细胞的单细胞研究路线,首先利用代谢标志物鉴定恶性胸腔积液中高代谢活性的肿瘤细胞,其次通过单细胞Sanger测序揭示这些肿瘤细胞具有一致的驱动基因突变特征,然后利用高通量测序技术对肿瘤细胞染色体拷贝数变异进行分析,从而揭示同一患者具有转移能力的肿瘤细胞即使具有相同的驱动基因突变特征,但在基因组层面上仍具有异质性,可进一步细分为若干子群。本研究结果对于更深入地理解肿瘤转移机制具有重要意义。
肺癌;胸腔积液;肿瘤细胞;单细胞测序
肺癌是威胁人类健康的主要疾病,其发病率及致死率在我国稳居各项恶性肿瘤之首[1]。对于同一种癌症,不同患者间的肿瘤具有很大的异质性[2,3];即使对于同一患者而言,由于基因组的不稳定,其恶性肿瘤在增殖过程中会产生不同的子代细胞亚群,它们在基因组[4]或表型[5]层面具有不同的分子与功能特征,不同的亚群其细胞增殖、侵袭转移以及耐药潜能方面都存在差异,尤其是存在一部分具有转移能力的肿瘤细胞能脱离原位组织并在远处器官形成转移灶[6],因此,肿瘤细胞的异质性为其发生转移和耐药提供了可能。但传统的肿瘤异质性研究主要基于不同位置原位组织样本中的群体细胞。虽然近年来迅速发展的高通量测序技术提高了肿瘤基因组解析的速度,但针对群体肿瘤细胞的基因组高通量测序不可避免的会引入大量非肿瘤细胞的遗传信息。有研究表明,从实体瘤组织内提取的肿瘤细胞,其中真正肿瘤细胞的DNA占总DNA的量不到50%,这样整体平均化的测序结果使得很多频率较低但非常重要的肿瘤基因组信息被湮没[7]。单细胞测序技术从根本上克服了这样的局限,它能够从单个细胞水平探究不同肿瘤细胞的基因组和转录组特征。Xu等[8]利用单细胞测序技术揭示了肾癌细胞单核苷酸突变特性;Hou 等[9]同样利用单细胞测序技术探究了骨髓增殖性肿瘤的发生发展机制,为肿瘤异质性研究提供了全新的思路。而Navin等[10]利用单细胞测序技术绘制了200多个肿瘤细胞的进化图谱,可见单细胞测序技术在肿瘤生物学方面有着极大的应用前景。
过去肿瘤异质性研究主要聚焦于肿瘤组织的异质性,比如对肿瘤组织的不同位置取样进行异质性分析,缺乏对具有转移能力的肿瘤细胞进行深入的分析。众所周知,转移是肿瘤致死的最重要因素,因此非常有必要对转移性的肿瘤细胞进行单细胞尺度上的分子分析,解析其异质性[11,12]。由于肺癌患者常伴有恶性胸腔积液的产生,胸腔积液中所含有的肿瘤细胞是肺癌胸膜转移的标志[13]。因此,肺癌患者恶性胸腔积液中的肿瘤细胞代表了从肿瘤组织脱落、具有转移能力的肿瘤细胞群体[14]。本研究以肺癌患者恶性胸腔积液中单个肿瘤细胞为研究对象,通过单细胞测序检测其基因组特征并分析这些肿瘤细胞在基因组层面上的异质性。由于胸腔积液中细胞成分复杂,包含多种类型的细胞,因此需首先使用本实验室前期开发的高代谢活性恶性肿瘤细胞的鉴定方法,并以多重置换扩增的单细胞基因组扩增技术和高通量测序技术为主要技术路线,建立一整套转移性肿瘤细胞的单细胞分析方案,从而进一步解析肿瘤细胞在分子层面的异质性,为更深入理解肿瘤转移机制提供更多信息。
1 材料与方法
1.1 样本收集
肺癌患者的胸腔积液样本收集自上海市胸科医院,5名患者均为非小细胞肺癌患者,临床分期均为Ⅳ期,其中男性患者3名,女性患者2名,样本收集及后续研究均通过伦理委员会审核,且在患者充分知情的情况下进行。
1.2 微孔阵列芯片制作及表面修饰
参照本实验室前期开发的微孔阵列芯片制作方法制作实验所需微孔阵列芯片[15]。将聚二甲基硅氧烷(polydimethylsiloxane, PDMS)的两种组分按照1∶10质量比充分搅拌混匀,真空抽去气泡,均匀倒于放置硅片模具的10 cm培养皿中。将皿置于80℃电热烘箱中烘烤2 h。将PDMS从模具上剥离,按照设计的边沿切割成具有400个小型数字编号区域,共计11万个直径为30 μm微孔的长方形芯片,然后与经等离子体表面处理的玻片紧密贴合,最后使用2%的Matrigel对PDMS芯片的微孔区域进行修饰。
1.3 高代谢活性肿瘤细胞的鉴定
临床收集的肺癌患者胸腔积液样本置于4℃条件下送至实验室。首先经100 μm孔径的细胞筛去除细胞聚团和杂质,然后在4℃条件下通过300离心5 min收集细胞沉淀,再加入一定量红细胞裂解液重悬细胞沉淀并室温避光裂解15 min。裂解结束后于4℃条件200离心5 min,用含0.1% BSA的HBSS重悬细胞,按照每1×107细胞加入20 μL CD45- APC抗体染色,室温翻转孵育,然后将细胞均匀铺至事先处理好的PDMS微孔芯片上并用无糖Dulbeccos Moddified Eagle Medium(DMEM)培养基洗去CD45-APC。在芯片上加入使用无糖DMEM培养基配制2-NBDG染色液,使其浓度达到0.4 mmol/L,37℃避光条件下孵育10 min,再用预冷的HBSS溶液快速清洗芯片,在低温下使用高内涵显微成像系统(ImageXpress. XLS. Molecular Devices公司, 美国)对细胞进行荧光成像。根据本实验室建立的方法鉴定芯片上的高代谢活性肿瘤细胞。
1.4 肿瘤单细胞回收与单细胞全基因组扩增
借助显微操作平台准确回收高代谢活性肿瘤单细胞,将肿瘤单细胞移入含有4 μL PBS的PCR管中。使用REPLI-g single cell kit (Qiagen, 德国)对单细胞全基因组进行扩增,具体实验操作参照试剂盒说明书。首先按照11∶1混合DLB与DTT配制单细胞裂解液,向肿瘤单细胞中加入3 μL细胞裂解液,65℃裂解10 min,加入3 μLStop solution终止裂解。按照表1配制单细胞基因组扩增反应液,向每个肿瘤单细胞样本中加入40 μL单细胞基因组扩增反应液,具体成分为29 μL REPLI-g sc Reaction Buffer、9 μL H2O、2 μL scPhi29 DNA polymerase。PCR仪中30℃孵育5 h;65℃,3 min使酶灭活。
1.5 单细胞全基因组扩增产物基因突变检测
针对表皮生长因子受体基因(epidermal growth factor receptor,)的18、19、20和21外显子分别设计对应特异性扩增引物用于基因突变检测[16](引物见表2)。使用2×Ex-Premix扩增目标片段,配制反应体系。具体PCR反应程序:94℃,3 min;94℃,15 s,60℃,20 s,72℃,30 s,进行30个循环,72℃,5 min。扩增产物经过凝胶电泳检验,有明显条带则证明扩增成功,剩余扩增产物进行Sanger测序。
1.6 单细胞全基因组测序
使用实验室设计的针对22条染色体特定片段的扩增引物对单细胞扩增产物进行质控,通过质检的单细胞进行后续建库测序。单细胞全基因组测序文库构建使用NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB,美国)。首先使用3 μL NEBNest dsDNA fragmentase在片段化缓冲液中37℃反应4 min,立即加入5 μL EDTA终止片段化;再使用0.65×恢复至室温的Agencourt AMPure XP beads吸附>500 bp的DNA片段并弃去,使用0.1×Agencourt AMPure XP beads吸附300~500 bp大小的片段,经现配的80%酒精洗涤两次后,用无核酸酶水洗脱建库DNA片段;使用Qubit dsDNA HS Assay Kit (Thermo, 美国)测定样本浓度;末端修复阶段使用样本量为60~100 ng,并在End Prep Enzyme Mix作用下,20℃孵育30 min,65℃孵育30 min;修复后的平末端加A及接头阶段,使用NEB Next Adaptor在Blunt/TA Ligase Master Mix及Ligation Enhancer的作用下,DNA平末端加上腺嘌呤及连接有胸腺嘧啶的特异性接头;为保证后续反应,需将U型环状特异性接头剪开形成双链,加入3 μL USER Enzyme在37℃条件下孵育15 min完成酶切;为纯化连上特异性接头的DNA片段,使用0.8×恢复至室温的Agencourt AMPure XP beads吸附目的DNA,使用现配的80%酒精洗去多余的接头,并用16 μL无核酸酶水室温孵育beads 2 min,置于磁力架吸附3 min,将上清移入0.2 mL PCR管中;全基因组测序文库预扩增阶段使用Q5 DNA Polymerase PreMix体系,针对每一个不同的单细胞全基因组文库引入不同的Index进行标记;文库扩增产物经Agencourt AMPure XP beads两步法纯化,使用现配的80%酒精洗涤beads两次,再用20 μL无核酸酶水洗脱文库DNA;最后使用Qubit dsDNA HS Assay Kit (Thermo, 美国)测定文库浓度,Agilent 2100 Bioanalyzer (Agilent, 美国)检测DNA片段分布,按照等摩尔质量混合不同样本文库,高通量测序使用HiSeq X Ten平台,按照PE150测序方案完成。

表1 单细胞基因组扩增反应液成分

表2 非小细胞肺癌EGFR基因检测扩增引物
1.7 数据质控与分析
测序数据质控采用MAPD值法。基于Multiple absolute pairwise difference算法:MAPD=Median (log2nCNRi+1-log2nCNRi),其中i代表单个窗口,MAPD值越大,噪音越大,数据质量越差。本研究采用0.45为阈值,去除MAPD值大于0.45的样本[17]。
以通过质控的单细胞下一代测序(Next generation sequencing, NGS)数据为分析单元,使用FASTQC工具评估数据质量,使用Trimmomatic-0.36程序[18]去除质量不好的碱基和多余的接头,采用处理PE测序模式数据的命令得到clean reads和数据处理日志。Clean reads获取原则为切除首端碱基质量小于3和末端碱基质量小于3的碱基,Windows的size为4个碱基,切除平均碱基质量小于15,舍弃小于36 bp的reads。针对每一个去除接头和低质量reads的单细胞测序数据,使用BWA比对软件[19](Burrows- Wheeler Alignment tool)将clean reads数据比对到UCSC hg19的参考基因组上并且输出SAM文件。为了后续计算,用Samtools工具[20]将SAM文件压缩成BAM文件,并进行染色体排序。为了提高计算速度,用Samtools对其建立索引,然后使用Picard (Picard-tools-1.119)将测序文库构建过程中由PCR预扩增引入的重复进行标记和删除,排除PCR过程造成的干扰。处理完成的每一个单细胞NGS数据,我们采用500 kb长度为窗口将其分割,记录每一个窗口内的reads数目,并对其进行GC含量的标准化。最后,采用R语言中DNAcopy package[21]环状二元分割法来计算染色体拷贝数,用于染色体拷贝数变异分析,最后用R语言画图可视化。
2 结果与分析
2.1 微孔阵列芯片设计
本研究采用本实验室前期设计的微孔阵列芯片,该芯片包含11万个直径为30 μm,深度为30 μm的微孔,分布于400个数字编号的方形区域中(图1,A和B)。微孔区域经过等离子修饰亲水,并滴加上400 μL PBS保持亲水性。
2.2 分选高代谢活性肿瘤单细胞
基于本实验室之前的研究,可通过荧光标记的葡萄糖类似物2-NBDG表征细胞的葡萄糖摄取能力,并将白细胞标志物CD45表达阴性,2-NBDG高摄取的细胞鉴定为高代谢活性的疑似肿瘤细胞,其恶性可通过单细胞测序进一步确认。图2A所示为2号患者胸腔积液中细胞成像结果,FITC通道强绿色荧光而APC通道无红色荧光(CD45-,NBDG+)即为CD45阴性的高代谢活性疑似肿瘤细胞,APC通道有红色荧光(CD45+)即表征白细胞,在5名患者胸腔积液中均发现此类疑似肿瘤细胞。基于芯片设计的可寻址功能,使用显微操作仪将目的细胞取出置于PBS中进行后续实验(图2B)。
图1 微孔阵列芯片实物图
A:芯片与干净的玻片粘合,置于培养皿中固定;B:编号为“227”的方形区域,细胞较均匀铺满微孔。
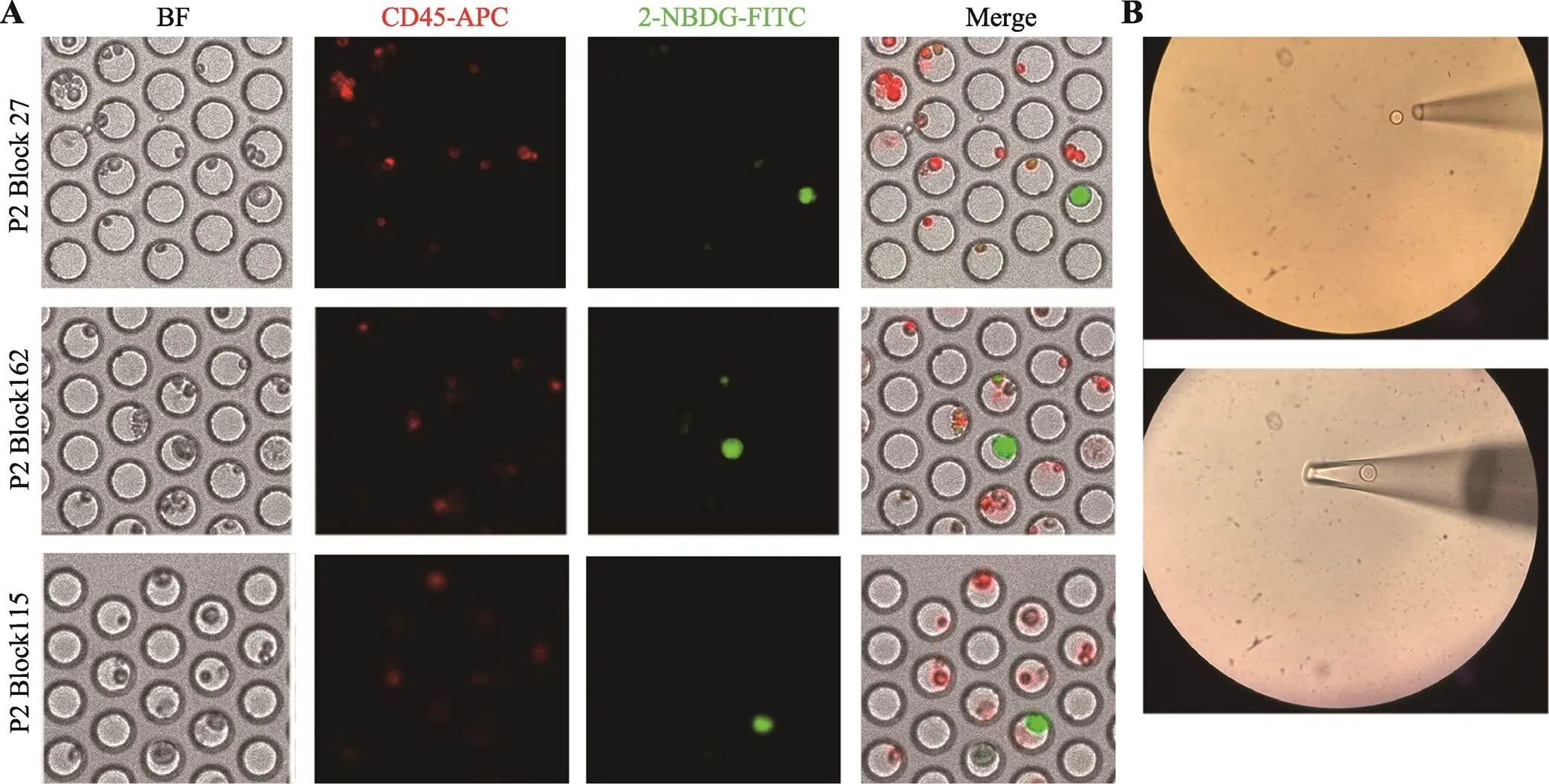
图2 胸腔积液中高代谢活性肿瘤细胞的鉴定及显微操作挑取单细胞示意图
A:2号患者3个典型的高代谢活性肿瘤细胞。BF表示明场下的成像图,CD45-APC表示在APC通道的成像图,2-NBDG-FITC表示在FITC通道的成像图,Merge表示将明场以及各荧光通道叠加之后的成像效果图;B:使用毛细玻璃管吸取和吹出目的细胞的示 意图。
2.3 单细胞全基因组扩增及Sanger测序
通过显微操作挑取的肿瘤单细胞进行全基因组扩增,扩增产物经过凝胶电泳确认单细胞扩增成功。由于非小细胞肺癌中负责信号转导的表皮生长因子受体发生突变导致其表达量升高,促进肿瘤的进展和转移,而针对突变的患者使用酪氨酸激酶抑制剂药物有着很好的效果,因此对其进行针对基因的Sanger测序可以判定其靶向治疗有效性,有着重大的临床意义[22~24]。结果显示:患者P2单细胞测序为-19Del (图3A),存在一个野生型(图3B),患者P1的肿瘤单细胞测序结果为-L858R突变(图3,C和D),患者P3和P5为-19Del,患者P4为-L858R突变,5名患者恶性胸腔积液中高代谢活性肿瘤单细胞Sanger测序结果与临床医生提供的患者突变信息吻合。同时,在同一个患者的单细胞样本中,用CD45−/2- NBDG+为标准筛选的高代谢活性肿瘤细胞,有一定比例的野生型,2号患者有1个单细胞是野生型,提示利用高代谢活性的标准筛选出的肿瘤细胞可能有着不存在驱动基因突变的亚群;P1、P3、,P4和P5号患者所有高代谢活性肿瘤单细胞均带有点突变。
2.4 高代谢活性肿瘤单细胞基因组拷贝数变异分析
对于通过22条染色体检测的单细胞样本(图4A),认为其全基因组扩增产物覆盖度较好适合进行后续拷贝数变异分析。肿瘤细胞的基因组极不稳定,容易发生基因组重排[25],且重排基因组的大小都在1 kb以上,亚显微结构上体现出扩增[26]或者缺失[27]的现象。基于实验室对单细胞建立的染色体拷贝数变异(copy number variants, CNV)分析路线,对每个患者的所有高代谢活性的肿瘤细胞都进行了CNV分析。结果表明,P2、P3和P5患者都是-19Del突变类型,但3者的CNV变异情况存在很大差异(图4,B~D)。P3患者的5个肿瘤细胞均存在驱动基因突变但是在基因组水平上没有大范围的变异情况可能属于早期转移的细胞,P2患者肿瘤细胞可细分为2个亚群,可能代表了转移能力不同的两个群体,P5患者存在较好的一致性,但其基因组变异谱与P2和P3均不同;P1患者的10个单细胞基因组拷贝数变异一致性好,大部分染色体都发生了扩增或者缺失,认为此类肿瘤细胞的突变比较多,恶性程度比较高(图4E);P4患者在驱动基因突变检测上有很好的一致性,但是从CNV水平看每个单细胞的拷贝数变异都有较大的异质性(图4F)。
3 讨论
肿瘤异质性是目前肿瘤治疗面临的最大难题,同一癌种在不同患者之间存在基因或表型的差异,且在一个患者肿瘤不同区域也存在分子层面的显著差异[28]。有研究表明,患者体内实体瘤本身就是由含有不同基因或表型特征的亚克隆细胞群体组成[29],它们在增殖速度、侵袭转移能力等方面都具有显著差异,当所处微环境[30,31]发生改变时,一部分不能适应的肿瘤细胞失巢凋亡[32],而另一部分则存活下来,更有一部分潜在转移性肿瘤细胞在外界不利因子的压力下,突破屏障进入到循环系统进而实现远端转移。自肿瘤异质性首次被提出之后,越来越多的临床医生开始关注肿瘤异质性与患者的治疗方案之间的关系。临床上对突变的晚期肺癌患者使用靶向药物可以杀死突变肿瘤细胞,但是通常患者会在几个月内表现出药物抵抗性,需不断更换治疗方案以延长其生存期,药物等外界环境刺激还可能会诱导体内部分转移潜能的肿瘤细胞从原位脱落实现远端转移。因此深入解析肿瘤异质性是解释肿瘤耐药、侵袭和转移机制的关键。
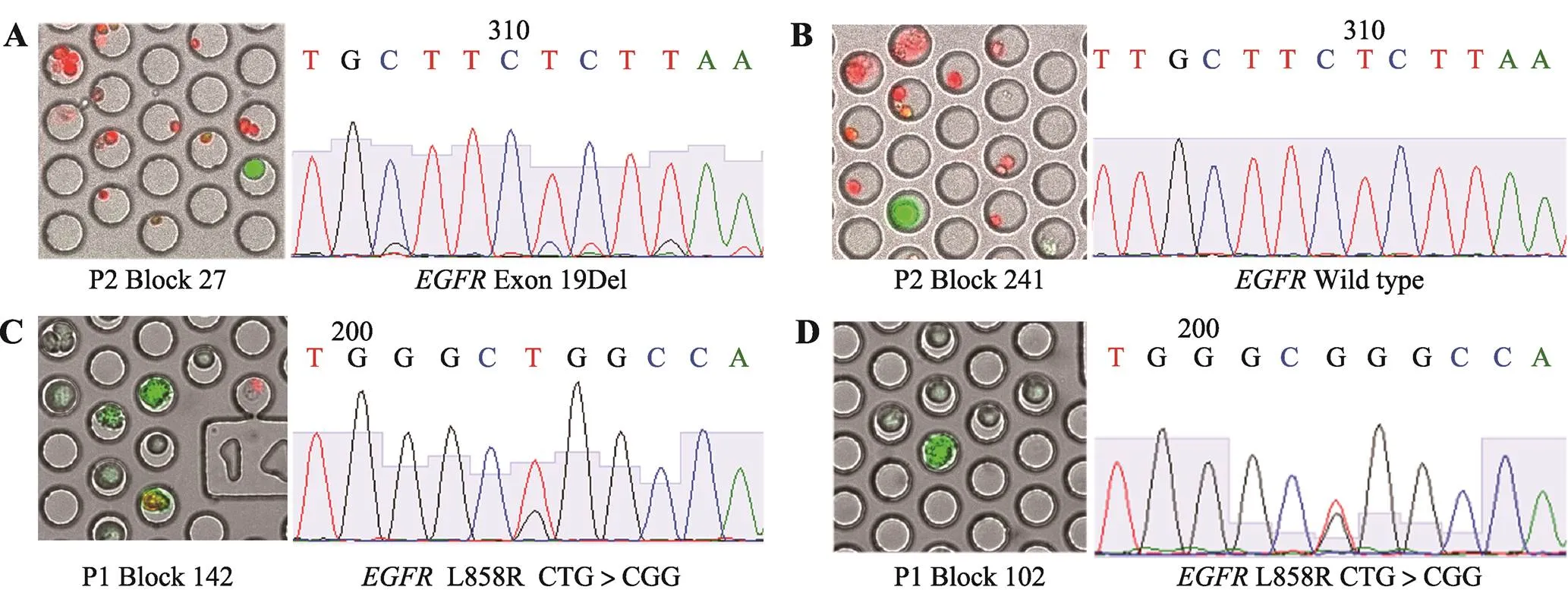
图3 肺癌患者恶性胸腔积液中高代谢活性肿瘤细胞Sanger测序结果
A:2号患者2-NBDG高摄取的单细胞,Sanger测序为基因19Del;B:2号患者2-NBDG高摄取的单细胞,Sanger测序为野生型;C,D:为1号患者2-NBDG高摄取的两个不同单细胞,Sanger测序均为基因21号外显子L858R点突变。
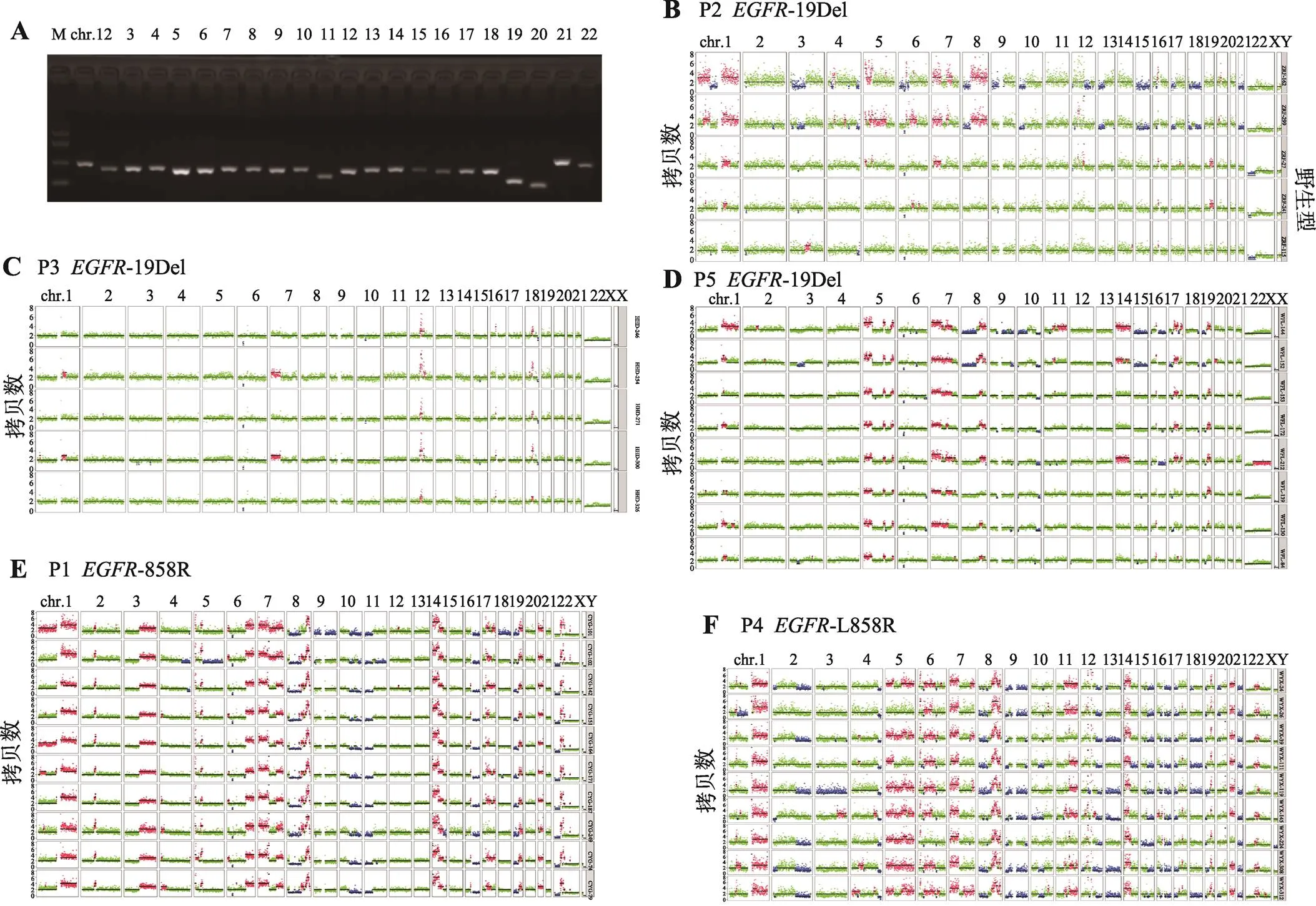
图4 单细胞全基因组质控及染色体拷贝数变异分析
A:单细胞全基因组扩增产物质量评估。泳道1:DNA marker;泳道2~23:分别表示22条染色体上特定基因片段的扩增产物;B:2号患者CNV结果,其突变类型为19Del;C:3号患者CNV结果,其突变类型为19Del;D:5号患者CNV结果,其突变类型为19Del;E:1号患者CNV结果,其突变类型为L858R;F:4号患者CNV结果,其突变类型为L858R。每一横排为一个单细胞的22对常染色体及一对性染色体拷贝数变异情况,每条染色体的绿色标注为正常的2倍体,红色标注为染色体拷贝数发生扩增,蓝色标注为染色体拷贝数发生缺失。
葡萄糖高摄取的肿瘤细胞代表了恶性胸腔积液中高代谢活性的细胞亚群[33],这部分细胞通过大量糖酵解方式获取能量[34,35]。本实验室前期研究认为它们是具有高转移潜能的细胞亚群,因此本研究以肺癌患者恶性胸腔积液中的高代谢活性细胞为研究对象对深入解析肿瘤异质性及转移机制有着重要意义。结果发现:突变类型均为-L858R的患者(P1和P4),同一个患者多个高代谢活性肿瘤细胞的拷贝数变异存在高度一致性,但是患者之间基因组水平上的拷贝数变异存在极大的差异,P1患者在染色体拷贝数扩增现象多于缺失,而P4患者的扩增和缺失现象都非常明显;同样突变类型均为-19Del的患者(P2、P3和P5),同一个患者的多个高代谢活性肿瘤细胞的拷贝数变异存在差异,可细分为若干亚群,患者之间的CNV变异类型也完全不一样,由此推测这样的差异可能是导致患者出现的不同转移和耐药情况的原因。针对转移性肿瘤细胞之前的研究主要有两种观点,其一是这种差异来自于原位灶本来就不同的细胞亚克隆[36],另一种观点认为是具有高度干性的肿瘤细胞亚群转移之后再形成的不同分子特点[37,38]。从同一个患者(P2和P5)的多个肿瘤细胞CNV结果看,可细分成不同的细胞亚群,虽然其突变类型一致,但是其基因组上明显的分群现象可能与肿瘤后期进展相关。恶性胸腔积液中高代谢活性肿瘤细胞应该同时具备这两种情况。本研究认为一些早期转移的患者(P3)其基因组水平变异是比较平的,显著区别于P1和P4高度变异的情况,这对于临床医生针对性治疗和最大程度使患者获益有极大的帮助。
综上所述,本研究建立了一整套针对肺癌患者恶性胸腔积液中转移性肿瘤细胞的单细胞分析方案。利用代谢标志物分选高代谢活性肿瘤细胞避免了胸腔积液中其他杂细胞的遗传信息干扰,从单细胞角度解析了同一患者多个转移性肿瘤细胞在基因组水平上的一致性,而相同驱动基因的患者之间存在显著差异,认为其差异可能导致患者在转移和耐药方面存在差异;而同一患者不同的细胞亚群可能是早期转移患者仍然存在潜在过渡态细胞群体。本文对于肿瘤异质性的研究尤其是对转移性肿瘤细胞的异质性研究将有望对肺癌患者临床治疗,耐药监测和复发转移提供更精确的生物学信息。
[1] Yu HX, Han ZG, Wang Y, Xin H. The clonal evolution and therapeutic approaches of lung cancer., 2014, 70(1): 63–71.
[2] Aparicio S, Mardis E. Tumor heterogeneity: next-generation sequencing enhances the view from the pathologist’s microscope., 2014, 15(9): 463.
[3] Swanton C. Intratumor Heterogeneity: evolution through Space and Time., 2012, 72(19): 4875–4882.
[4] Wang LW, Wang L, Li N. Clinical cancer genomics and personalized treatment., 2010, 15(6): 481–486.王理伟, 王雷, 李宁. 临床肿瘤基因组学与肿瘤个体化治疗. 临床肿瘤学杂志, 2010, 15(6): 481–486.
[5] Peng QP, Liang HJ. Specific phenotype for glycolysis in malignant tumor cells and its significance.l, 2009, 14(5): 470–473.彭秋平, 梁后杰. 糖酵解代谢在恶性肿瘤细胞中的特异性表型及其意义. 临床肿瘤学杂志, 2009, 14(5): 470– 473.
[6] Khoo BL, Grenci G, Lin YB, Lee SC, Han J, Lim CT. Expansion of patient-derived circulating tumor cells from liquid biopsies using a CTC microfluidic culture device., 2018, 13(1): 34–58
[7] Gerlinger M, Rowan A, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing., 2012, 366(10): 883–892.
[8] Xu X, Hou Y, Yin XY, Bao L, Tang AF, Song LT, Li FQ, Tsang S, Wu K, Wu HJ, He WM, Zeng L, Xing MJ, Wu RH, Jiang H, Liu X, Cao DD, Guo GW, Hu XD, Gui YT, Li ZS, Xie WY, Sun XJ, Shi M, Cai ZM, Wang B, Zhong MM, Li JX, Lu ZH, Gu N, Zhang XQ, Goodman L, Bolund L, Wang J, Yang HM, Kristiansen K, Dean M, Li YR, Wang J. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor., 2012, 148(5): 886–895.
[9] Hou Y, Song LT, Zhu P, Zhang B, Tao Y, Xu X, Li FQ, Wu K, Liang J, Shao D, Wu HJ, Ye XF, Ye C, Wu RH, Jian M, Chen Y, Xie W, Zhang RR, Chen L, Liu X, Yao XT, Zheng HC, Yu C, Li QB, Gong ZL, Mao M, Yang X, Yang L, Li JX, Wang W, Lu ZH, Gu N, Wang J. Single-Cell exome sequencing and monoclonal evolution of a-Negative myeloproliferative neoplasm., 2012, 148(5): 873– 885.
[10] Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. Tumour evolution inferred by single-cell sequencing., 2011, 472(7341): 90–94.
[11] Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms., 2011, 147(2): 275– 292.
[12] Yang YL, Chu JY, Wang MR. Tumor genetic heterogeneity.,2013, 35(1): 1–9.杨壹羚, 褚嘉祐,王明荣. 肿瘤遗传异质性. 遗传, 2013, 35(1): 1–9.
[13] Fenton KN, Richardson JD. Diagnosis and management of malignant pleural effusions., 1995, 170(1): 69–74.
[14] Roberts ME, Neville E, Berrisford RG, Antunes G, Ali NJ, BTS Pleural Disease Guideline Group. Management of a malignant pleural effusion: British thoracic society pleural disease guideline 2010., 2010, 65 (Suppl. 2): ii32– ii40.
[15] Tang Y, Wang Z, Li ZM, Kim J, Deng YL, Li Y, Heath JR, Wei W, Lu S, Shi QH. High-throughput screening of rare metabolically active tumor cells in pleural effusion and peripheral blood of lung cancer patients., 2017, 114 (10): 2544–2549.
[16] Sun S, Deng YL. Single-cell detection of EGFR gene mutation in circulating tumor cells in lung cancer., 2015, 37(12): 1251–1257.
孙帅, 邓宇亮. 肺癌循环肿瘤细胞的单细胞EGFR基因突变检测. 遗传, 2015, 37(12): 1251–1257.
[17] Cai X, Evrony GD, Lehmann HS, Elhosary PC, Mehta BK, Poduri A, Walsh CA. Single-Cell, Genome-wide sequencing identifies clonal somatic Copy-Number variation in the human brain., 2015, 10(4): 645.
[18] Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data., 2014, 30(15): 2114–2120.
[19] Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform., 2009, 25(14): 1754–1760.
[20] Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/ map format and samtools., 2009, 25(16): 2078–2079.
[21] Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data., 2007, 23(6): 657–663.
[22] Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR., 2010, 362(25): 2380–2388.
[23] Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, Ludovini V, Magrini E, Gregorc V, Doglioni C, Sidoni A, Tonato M, Franklin WA, Crino L, Bunn PA, Varella-Garcia M. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer., 2005, 97(9): 643–655.
[24] Asahina H, Yamazaki K, Kinoshita I, Sukoh N, Harada M, Yokouchi H, Ishida T, Ogura S, Kojima T, Okamoto Y, Fujita Y, Dosaka-Akita H, Isobe H, Nishimura M. A phase II trial of gefitinib as first-line therapy for advanced non-small cell lung cancer with epidermal growth factor receptor mutations., 2006, 95(8): 998–1004.
[25] Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders., 2002, 18(2): 74–82.
[26] Litt M, Luty JA. A hypervariable microsatellite revealed byamplification of a dinucleotide repeat within the cardiac muscle actin gene., 1989, 44(3): 397–401.
[27] Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters., 2006, 103(5): 1412–1417.
[28] Wang DC, Wang W, Zhu B, Wang X. Lung cancer heterogeneity and new strategies for drug therapy., 2017, 58: 531–546.
[29] Greaves M, Maley CC. Clonal evolution in cancer., 2012, 481(7381): 306–313.
[30] He YL, Ying Y, Xu YL, Su JF, Luo H, Wang HF. Effects of Lycium barbarum polysaccharide on tumor microenvironment T-lymphocyte subsets and dendritic cells in H22-bearing mice., 2005, 3(5): 374–377.
何彦丽, 应逸, 许艳丽, 苏俊芳, 罗惠, 王惠峰. 枸杞多糖对荷瘤小鼠肿瘤微环境T淋巴细胞亚群及树突状细胞的影响. 中西医结合学报, 2005, 3(5): 374–377.
[31] Liotta LA, Kohn EC. The microenvironment of the tumour-host interface., 2001, 411(6835): 375–379.
[32] Douma S, van Laar T, Zevenhoven J, Meuwissen R, van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB., 2004, 430(7003): 1034–1039.
[33] Pantel K, Alix-panabières C. Functional studies on viable circulating tumor cells., 2015, 62(2): 328–334.
[34] Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review., 1989, 49(23): 6449– 6465.
[35] Kalmus J, Okunieff P, Vaupel P. Effect of intraperitoneal versus intravenous glucose administration on laser Doppler flow in murine FSaII tumors and normal skin., 1989, 49(22): 6313–6317.
[36] Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution., 2009, 138(5): 822–829.
[37] Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors., 2003, 63(18): 5821– 5828.
[38] Heidt DG, Li C, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells., 2007, 67(3): 1030–1037.
Identification and single-cell sequencing analysis of rare tumor cells in malignant pleural effusion of lung cancer patients
Baojun Wu, Zhuo Wang, Yu Dong, Yuliang Deng, Qihui Shi
Tumor heterogeneity refers to distinct genomic or phenotypic characteristics of tumor cells. Under the environmental or drug stress, tumor cells exhibit different responses, corresponding to different properties of cell proliferation, invasion, metastasis and drug resistance. In particular, a small fraction of tumor cells are capable of detaching from primary tumor sites and initiating distant metastases. Thus, tumor heterogeneity sets the basis for tumor resistance and metastasis. Traditional methods in studying tumor heterogeneity are mainly based on bulk cells from different locations in primary tumors, lacking analysis at the single-cell level and of metastatic tumor cells. This study establishes a single-cell method to study metastatic tumor cells in malignant pleural effusions of lung cancer patients. Metabolically active tumor cells in malignant pleural effusions are firstly identified with a metabolic marker 2-NBDG, a fluorescent glucose analog. These metabolically active tumor cells are confirmed to harbor the same driver oncogenic mutations by Sanger sequencing, followed by high-throughput sequencing to analyze copy number variation profiles. Our results show metastatic tumor cells in pleural effusion have the same driver mutations but different features in copy number variation patterns. The study provides new insights to understand the mechanism of tumor metastasis.
lung cancer; pleural effusion; tumor cells; single-cell sequencing
2018-10-31;
2018-12-27
国家自然科学基金项目(编号:21775103, 81501613)和上海交通大学“医工交叉基金”项目(编号:YG2017QN54)资助[Supported by the National Natural Science Foundation of China (Nos. 21775103, 81501613) and Medical Engineering Cross Research Fund of Shanghai Jiao Tong University(No. YG2017QN54)]
吴保军,硕士研究生,专业方向:循环肿瘤细胞单细胞测序。E-mail: wbaojun@sjtu.edu.cn
施奇惠,教授,研究方向:液体活检与单细胞测序。E-mail: qihuishi@sjtu.edu.cn
10.16288/j.yczz.18-230
2019/1/14 13:15:26
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190114.1315.008.html
(责任编委: 周钢桥)
