基于基因家族大小的比较研究脊椎动物的适应性进化
2019-02-28孟玉杨若林
孟玉,杨若林
基于基因家族大小的比较研究脊椎动物的适应性进化
孟玉,杨若林
西北农林科技大学生命科学学院,杨凌 712100
同源基因家族的拷贝数在不同物种间普遍存在差异,这种差异是由不同的基因得失速率引起。众所周知,基因拷贝数变异是特定物种表型创新的可能原因。本研究选取具有代表性的脊椎动物主要类群并跨约6亿年进化时间的64个物种,鉴定了它们的同源基因家族,揭示了脊椎动物基因家族大小的进化模式。结果表明:在推断的存在于脊椎动物最近共同祖先的6857个基因家族中,有6712个都在至少一个种系中发生了大小的变化,而且基因家族在大多数种系中都是收缩的;其中,霍氏树懒()中有最高的基因家族收缩水平,而在斑马鱼()中则相反。基于脊椎动物基因家族大小进化的高度动态性,本研究从基因家族大小变化的角度鉴定了一些可能与特定脊椎动物类群进化有关的基因组信号。结果观察到在现存真骨鱼类最近共同祖先基因组中出现了可能因全基因组复制所导致的高比例的基因家族扩增现象,随后在后裔物种中发生基因收缩事件。此外,本研究还发现了硬骨鱼特异性的基因可能对这些鱼类在水生环境中的适应性进化有所贡献的证据,如在有些硬骨鱼中基因与鳍、尾巴、肾脏等发育有关。本研究结果有助于深入了解脊椎动物基因家族大小的进化,同时为理解脊椎动物基因组进化与表型多样性的联系提供了理论证据。
脊椎动物;基因家族;适应性进化;基因
脊椎动物亚门是脊索动物门中物种数量最多、结构最复杂的一个亚门,大约在5~6亿年前从其他脊索动物(头索动物和尾索动物)中分歧出来[1,2],并演化出无颌类、鱼类、两栖类、爬行类、鸟类和哺乳类,经历了成功的演化革新和适应。鉴定出脊椎动物间表型差异背后潜在的遗传变化,并确定导致这种变化的进化动力虽然具有挑战性,但有着深刻的科学意义。
基因复制是新基因产生及基因家族扩增的主要机制之一[3],为生物体表型的创新及多样化等提供了遗传基础[4],并且与生物体基因组大小的进化和物种分化等紧密相关[5]。与基因复制相比,基因丢失曾被认为仅与冗余的基因拷贝的丢失有关,而不会产生明显的功能影响,因此常被忽视。然而,与日俱增的基因组学数据揭示了基因丢失作为遗传变异的普遍来源,其具有引起适应性表型多样性的巨大潜能,是一种非常重要的进化动力[6]。如虎尾海马()基因组中的基因扩增和丢失与其特殊形态的演化密切相关。Lin等[7]对虎尾海马基因组进行了测序与分析,发现该物种基因组中Pastn (patristacin)基因家族(一种虾红素金属蛋白酶基因家族)经历了扩增,这与海马雄性孕育这一独特的繁殖方式密切相关。此外,虎尾海马基因组中(proline/glutamine-rich secretory calcium- binding phosphoprotein)基因和基因的丢失分别是导致其没有牙齿和腹鳍的重要原因。
通过全基因组比较分析,已经揭示了不同物种间许多基因家族拷贝数发生了显著的数量变化[8~11],这种变化与基因得失速率息息相关,且受到自然选择与遗传漂变的共同作用[6,12,13]。物种间表型的差异与基因家族大小的差异关系密切,如抗冻糖蛋白(antifreeze glycoprotein,)基因在南极鱼亚目鱼类基因组中发生了大量扩增,在南极鱼类适应低温环境中发挥了非常重要的作用[14]。除基因外,铁调素、卵壳蛋白等100多个参与低温适应相关生物学途径的基因也在南极鱼类进化中发生了显著扩增[15],这体现出特定基因拷贝数的增加是南极鱼类适应持续寒冷环境的一种机制。研究表明,不同种系间基因家族大小的变化可能与物种形成或适应性有重要联系[16~18]。例如,Yu等[18]对非人灵长类高海拔适应机制的研究中发现,与恒河猴()相比,生活在海拔高度为3500~4500米的滇金丝猴()基因组中有1187个基因家族发生了扩增,对其中231个显著扩增基因家族进行的功能富集分析表明,这些基因主要参与DNA修复和损伤应答以及氧化磷酸化过程。这一结果被认为可能与滇金丝猴暴露于高的紫外线辐射以及高海拔生存所需的能量代谢速率的增加有关。
植物和动物中基因家族大小的进化模式均已被广泛研究。然而,许多研究往往只涉及少数物种或只关注一个或某些基因家族的进化[19~22],缺乏全基因组水平的大规模分析。近年来,随着测序技术的发展,超过100种脊椎动物的全基因组已经被测序完成[23],这些数据的获得为人们揭示以下生物学问题提供了契机:(1)在脊椎动物中,物种间大规模的基因组差异,如基因家族大小的显著变化是否在物 种的适应性进化中起到了重要的作用;(2)物种或种系特异性基因的特征(包括表达模式和功能等)能否在一定程度上反映出物种间表型的差异。为了回答上述问题,本文选取64个涵盖了脊椎动物几乎所有类群(无颌类、鱼类、两栖类、爬行类、鸟类和哺乳类)的物种作为研究对象,从大的进化时间跨度上揭示了脊椎动物基因家族的扩增、收缩模式;并结合表达数据和功能注释评估了物种或种系特异性基因对物种特有表型的影响。本研究为深入了解脊椎动物基因家族大小的进化、理解脊椎动物间的基因组差异和表型多样性提供了新的见解。
1 材料与方法
1.1 数据来源
64个脊椎动物物种及2个外群物种——玻璃海鞘()和萨氏海鞘()完整的蛋白质组数据均下载自Ensembl v.84数据库。64个脊椎动物物种包含了1种无颌纲物种、12种鱼类、1种两栖动物、2种爬行动物、5种鸟类及43种哺乳动物。其中哺乳动物包括1种单孔目、3种有袋目、2种贫齿目、3种非洲兽总目、14种劳亚兽总目、2种兔形目、5种啮齿目、1种树鼩目和12种灵长目(表1)。从Ensembl网站(http://mar2016. archive. ensembl.org/info/about/speciestree.html)获取了这66个物种的系统发生关系。
1.2 物种间直系同源基因的鉴定
为了获得高质量的蛋白质序列数据用以鉴定基因的同源关系,对上述66个物种的蛋白质组数据按以下两个条件进行过滤:(1)去除长度小于50个氨基酸的蛋白质;(2)对于由可变剪切产生的多个转录本所翻译的蛋白质,只保留每个基因最长转录本对应的蛋白质。过滤之后,66个物种共1 149 492条蛋白质序列作为输入数据提交至OrthoMCL v2.0.9[24]进行蛋白聚类。该软件运行中的两个关键步骤是:(1) All-against-all BLASTP,即使用BlastP v2.2.31将每个蛋白与所有其他蛋白进行比对(-value < 1× 10–6),产生原始的blast输出;(2)使用马尔科夫聚类算法(Markov cluster algorithm, MCL)对解析的Blast结果构建马尔科夫矩阵,然后产生最终的基因家族[25]。MCL聚类的重要参数膨胀系数设为1.5。
1.3 基因家族大小分析
将每个物种的所有基因家族按其拷贝数分为3类:(1)单拷贝基因家族,每个家族包含的基因数目为1,即通常所说的单拷贝基因;(2)包含两个拷贝的基因家族,即双拷贝基因家族;(3)包含3个及3个以上拷贝的多拷贝基因家族。
本研究中每一个物种的Orphan基因家族(或基因)都是与其他65个物种进行比较得到的。例如,以人()为例,当某基因在除人以外的所有其他65个物种中都没有与之对应的同源基因时,就说明该基因是人的基因。
1.4 基因得失的似然法分析
CAFE (computational analysis of gene family evolution, version 3.0)是研究基因家族大小进化的统计分析工具,使用生灭模型对基因家族大小在特定系统发生树上的进化过程进行建模,并确定出各个分支上基因家族的扩增和收缩模式[26]。
由于上述66个物种基于分子水平的系统发生树与取自TimeTree[27]标有分歧时间的系统发生树不完全一致,为了便于分析和保证数据的可靠性,本研究从中选取57个脊椎动物物种进行后续分析。输入CAFE软件的树文件,为所选57个物种的Newick格式的有根系统发育树,且分支长度代表物种的分歧时间。数据文件是相应这些物种的各个基因家族大小的数据。使用的软件参数为:-p 0.05 -r 1000 -filter。最后通过lambda -s估算出所有基因家族总体的生灭参数λ。对于进化速率显著高于(<0.0001)全基因组平均值的基因家族[28],该软件使用Viterbi法识别出相应的分支,即基因家族大小发生显著变化(< 0.005)的分支[29]。
λ是基因家族大小进化分析中的一个重要参数,被用来度量单位时间(每百万年)内每个基因的得失概率。本研究中λ的估计值为0.0006,代表了所有基因家族整体水平的最有可能的生灭速率,或者说是基因家族随时间推移而扩增或收缩的速率。举例来说,所评估的基因得失速率意味着在特定基因组(如人类基因组)中,每百万年大约有13.467个新的拷贝和13.467个新的丢失被固定(0.0006得失/基因/百万年× 22 445基因)。
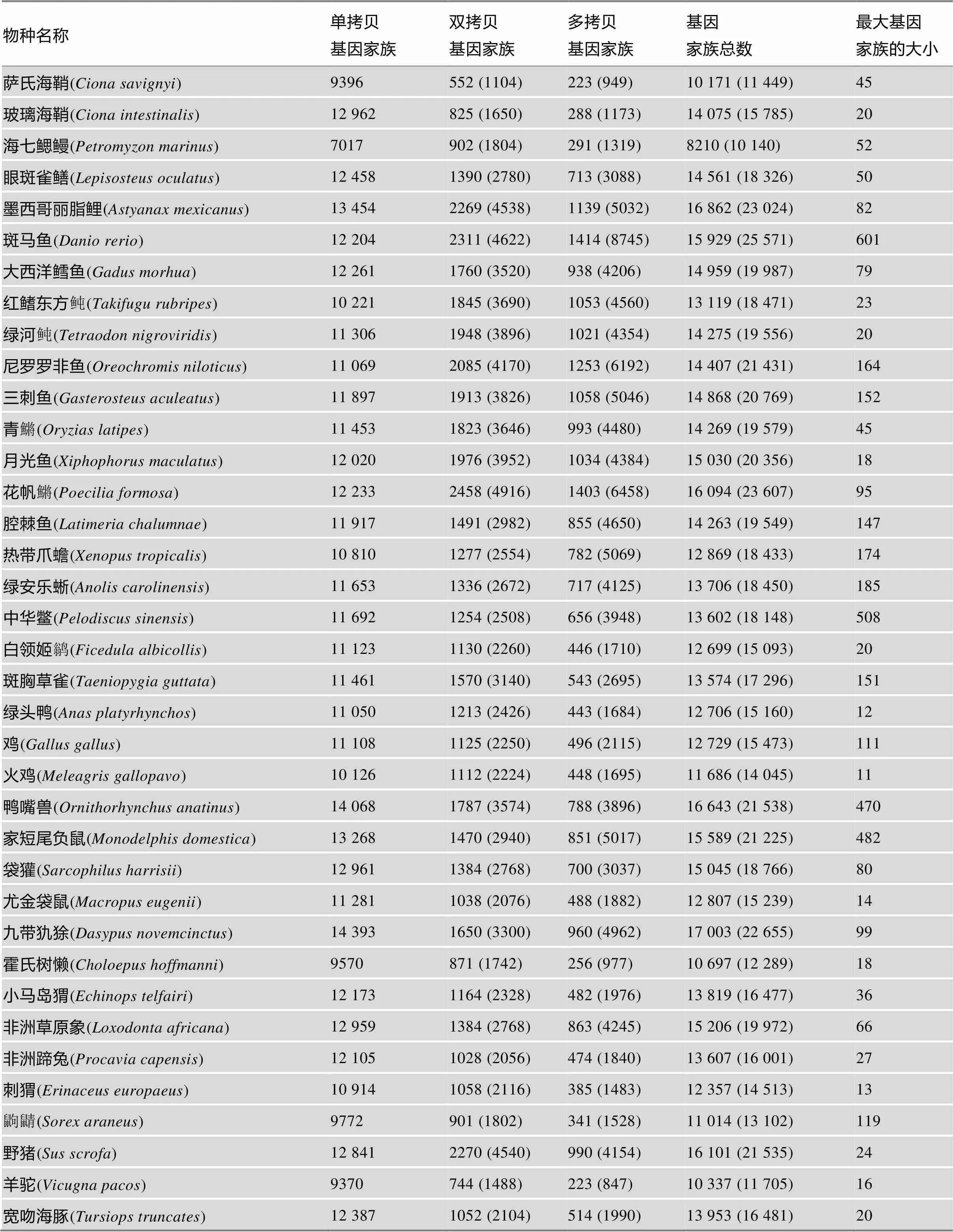
表1 66个物种基因家族及成员基因数量
续表
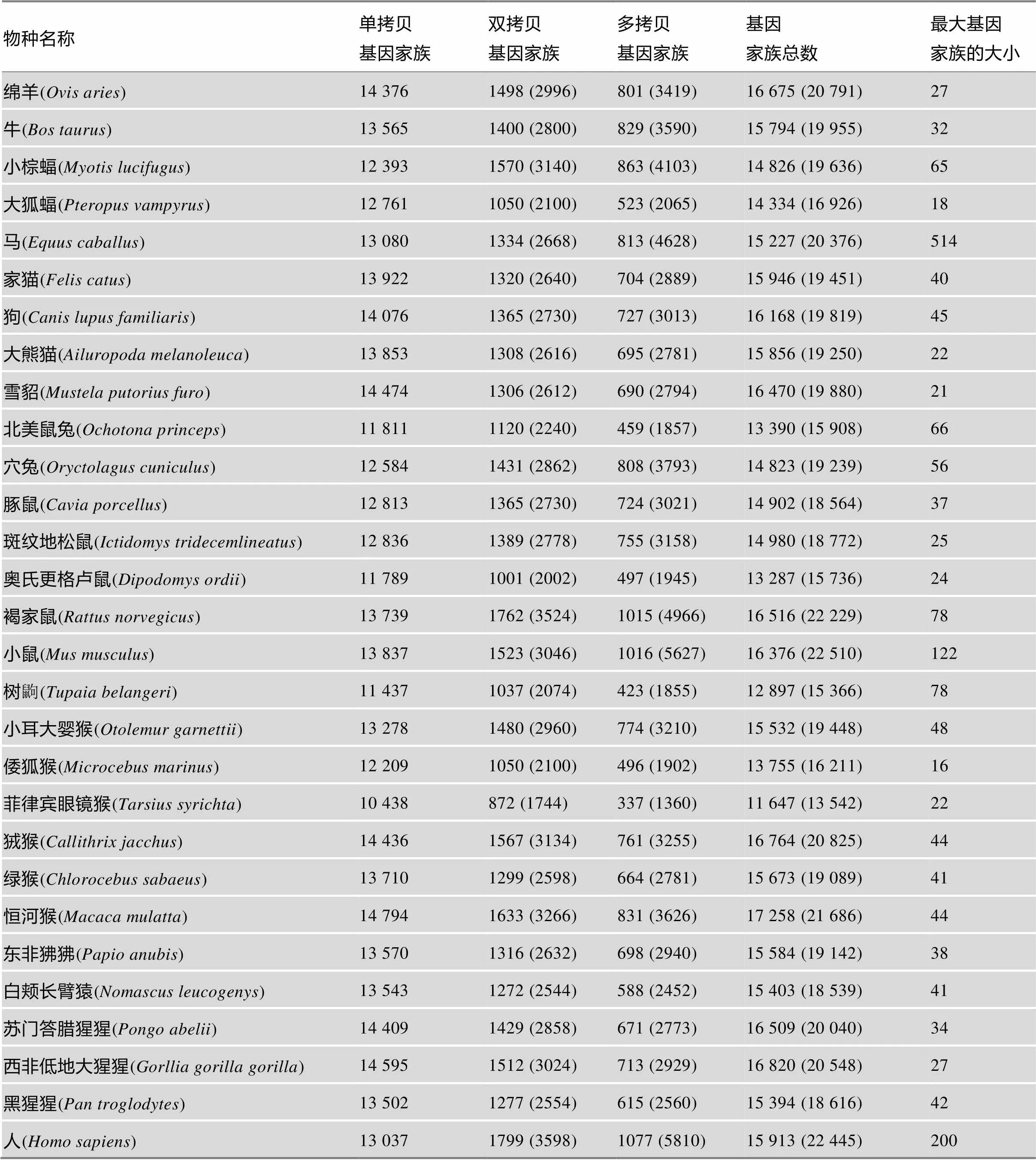
物种名称单拷贝基因家族双拷贝基因家族多拷贝基因家族基因家族总数最大基因家族的大小 绵羊(Ovis aries)14 3761498 (2996)801 (3419)16 675 (20 791)27 牛(Bos taurus)13 5651400 (2800)829 (3590)15 794 (19 955)32 小棕蝠(Myotis lucifugus)12 3931570 (3140)863 (4103)14 826 (19 636)65 大狐蝠(Pteropus vampyrus)12 7611050 (2100)523 (2065)14 334 (16 926)18 马(Equus caballus)13 0801334 (2668)813 (4628)15 227 (20 376)514 家猫(Felis catus)13 9221320 (2640)704 (2889)15 946 (19 451)40 狗(Canis lupus familiaris)14 0761365 (2730)727 (3013)16 168 (19 819)45 大熊猫(Ailuropoda melanoleuca)13 8531308 (2616)695 (2781)15 856 (19 250)22 雪貂(Mustela putorius furo)14 4741306 (2612)690 (2794)16 470 (19 880)21 北美鼠兔(Ochotona princeps)11 8111120 (2240)459 (1857)13 390 (15 908)66 穴兔(Oryctolagus cuniculus)12 5841431 (2862)808 (3793)14 823 (19 239)56 豚鼠(Cavia porcellus)12 8131365 (2730)724 (3021)14 902 (18 564)37 斑纹地松鼠(Ictidomys tridecemlineatus)12 8361389 (2778)755 (3158)14 980 (18 772)25 奥氏更格卢鼠(Dipodomys ordii)11 7891001 (2002)497 (1945)13 287 (15 736)24 褐家鼠(Rattus norvegicus)13 7391762 (3524)1015 (4966)16 516 (22 229)78 小鼠(Mus musculus)13 8371523 (3046)1016 (5627)16 376 (22 510)122 树鼩(Tupaia belangeri)11 4371037 (2074)423 (1855)12 897 (15 366)78 小耳大婴猴(Otolemur garnettii)13 2781480 (2960)774 (3210)15 532 (19 448)48 倭狐猴(Microcebus marinus)12 2091050 (2100)496 (1902)13 755 (16 211)16 菲律宾眼镜猴(Tarsius syrichta)10 438872 (1744)337 (1360)11 647 (13 542)22 狨猴(Callithrix jacchus)14 4361567 (3134)761 (3255)16 764 (20 825)44 绿猴(Chlorocebus sabaeus)13 7101299 (2598)664 (2781)15 673 (19 089)41 恒河猴(Macaca mulatta)14 7941633 (3266)831 (3626)17 258 (21 686)44 东非狒狒(Papio anubis)13 5701316 (2632)698 (2940)15 584 (19 142)38 白颊长臂猿(Nomascus leucogenys)13 5431272 (2544)588 (2452)15 403 (18 539)41 苏门答腊猩猩(Pongo abelii)14 4091429 (2858)671 (2773)16 509 (20 040)34 西非低地大猩猩(Gorllia gorilla gorilla)14 5951512 (3024)713 (2929)16 820 (20 548)27 黑猩猩(Pan troglodytes)13 5021277 (2554)615 (2560)15 394 (18 616)42 人(Homo sapiens)13 0371799 (3598)1077 (5810)15 913 (22 445)200
括号中的数字表示成员基因的数量;基因家族的大小指基因家族中包含的基因个数。
1.5 基因表达和GO (Gene Ontology)注释
从Expression Atlas数据库分别下载了人的16种组织(肝脏、淋巴结、甲状腺、骨骼肌、前列腺、大脑、睾丸、肾脏、肾上腺、肺脏、白细胞、卵巢、脂肪、乳腺、结肠和心脏)、鸡()的9种组织(大脑、心脏、肝脏、脾脏、肺脏、肾脏、结肠、睾丸和骨骼肌)的基因表达数据。斑马鱼的12种组织(骨、大脑、胚胎、卵巢、心脏、肠、肾脏、肝脏、肌肉、成熟卵泡、鳃和睾丸)的基因表达数据下载自Bgee数据库。基因表达的组织特异性参照文献[30,31]中描述的组织特异性指数来表示,计算公式如下:
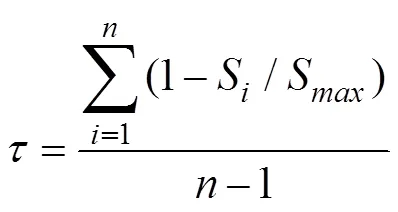
其中,是组织的数量,S是基因在第个组织中的表达量,S代表基因在所有组织中的最大表达量。本研究将≥0.85的基因视为组织特异性表达的基因,并关注这类基因最大表达值对应的组织;使用GOSlim对感兴趣的基因集进行功能富集分析。
2 结果与分析
2.1 基因家族大小的跨物种分布模式
为了鉴定脊椎动物间的直系同源基因,使用OrthoMCL[24]对涵盖了无颌类、鱼类、两栖类、爬行类、鸟类、哺乳类的64个脊椎动物物种和2个海鞘纲尾索动物物种(表1,图1A)共1 149 492个蛋白质序列进行了聚类分析,共产生32 498个直系同源基因家族。其中1648个基因家族是所有64个脊椎动物物种所共有,这可能代表了脊椎动物“核心”蛋白质组。
本研究首先对每个物种基因组中3类基因家族及其成员基因的数量进行了统计。在所研究的物种中,基因家族总数从8210 (海七鳃鳗,)至17 258 (恒河猴)不等(表1)。每个物种最大的基因家族由11 (火鸡,)至601 (斑马鱼)个基因组成(表1),这显示基因家族大小有着较大的跨物种变异程度。
进一步统计显示,除斑马鱼外,脊椎动物各物种基因组中半数以上的基因都以单拷贝的形式存在(图1B)。与双拷贝基因相比,单拷贝和多拷贝基因在各物种基因组中所占比例有更大差异。具体而言,双拷贝基因家族中的基因数占各物种总基因数的比例从12.4% (大狐蝠)至21.1% (野猪)不等,斑马鱼基因组中有最多的多拷贝基因(34.2%)和最少的单拷贝基因(47.7%),而羊驼基因组中有最少的多拷贝基因(7.2%)和最多的单拷贝基因(80.1%) (图1B)。
2.2 基因家族的扩增与收缩
基因得失的似然法分析中,需要假定所分析的基因家族在所有物种最近共同祖先中至少含有一个基因。在57个脊椎动物物种包含的28 084个基因家族中,只有6857个基因家族符合这一要求,因此本研究只对这些基因家族的扩增与收缩模式进行分析(图2)。
脊椎动物最近共同祖先处6857个基因家族中有6712个都在至少一个种系中发生了扩增或收缩。在57个脊椎动物物种组成的系统发育树的不同分支上基因家族扩增和收缩的模式来看,脊椎动物基因家族在大部分种系中都是收缩的,其中霍氏树懒中有最大程度的收缩(发生扩增和收缩的基因家族分别有74个和2151个),而斑马鱼中有最大程度的扩增(发生扩增和收缩的基因家族分别有912个和343个) (图2)。在鸟类中,除了斑胸草雀这一末端分支上发生了相对多的基因家族扩增以外,其他鸟类的基因家族均发生了较大收缩,这与鸟类基因组进化过程中整体的基因组变小现象一致[32]。已知鸟类基因组是羊膜动物中最小的,研究表明广泛的基因丢失比转座子活性降低对维持鸟类较小的基因组有更重要的贡献[33]。在辐鳍鱼中,真骨附类进化早期有大量的基因家族发生扩增,随后又有较多的基因家族呈现出收缩的模式(图2),这与真骨附类祖先物种发生了特有的全基因组复制以及复制后往往伴随着大量的基因丢失现象基本吻合[34,35]。
似然法分析能够识别基因家族大小的进化速率显著高于全基因组平均值的基因家族[28]。在所分析的6857个基因家族中,有148个是快速进化的基因家族(<0.01%),其中22个快速进化的基因家族在人这一末端分支上发生了显著扩增。例如,CT抗原中CTAGE (cutaneous T-cell-lymphoma-associated antigen)基因家族是一类由生殖细胞系基因编码的肿瘤/睾丸抗原,在人类的很多肿瘤中CT抗原会异常表达[36]。本研究的数据显示,该基因家族在人类基因组中有10个拷贝,而在黑猩猩中的拷贝数为2,用CAFE软件所推断的人与黑猩猩最近共同祖先中的该基因家族拷贝数为2。之前有研究发现CTAGE基因家族在灵长类的进化中发生了快速的扩增,人类基因组中的CTAGE基因家族包含了多个单外显子基因拷贝,这些单外显子拷贝基因受到明显的正选择作用,有可能对人类早期进化中适应性表型的产生有贡献[37]。
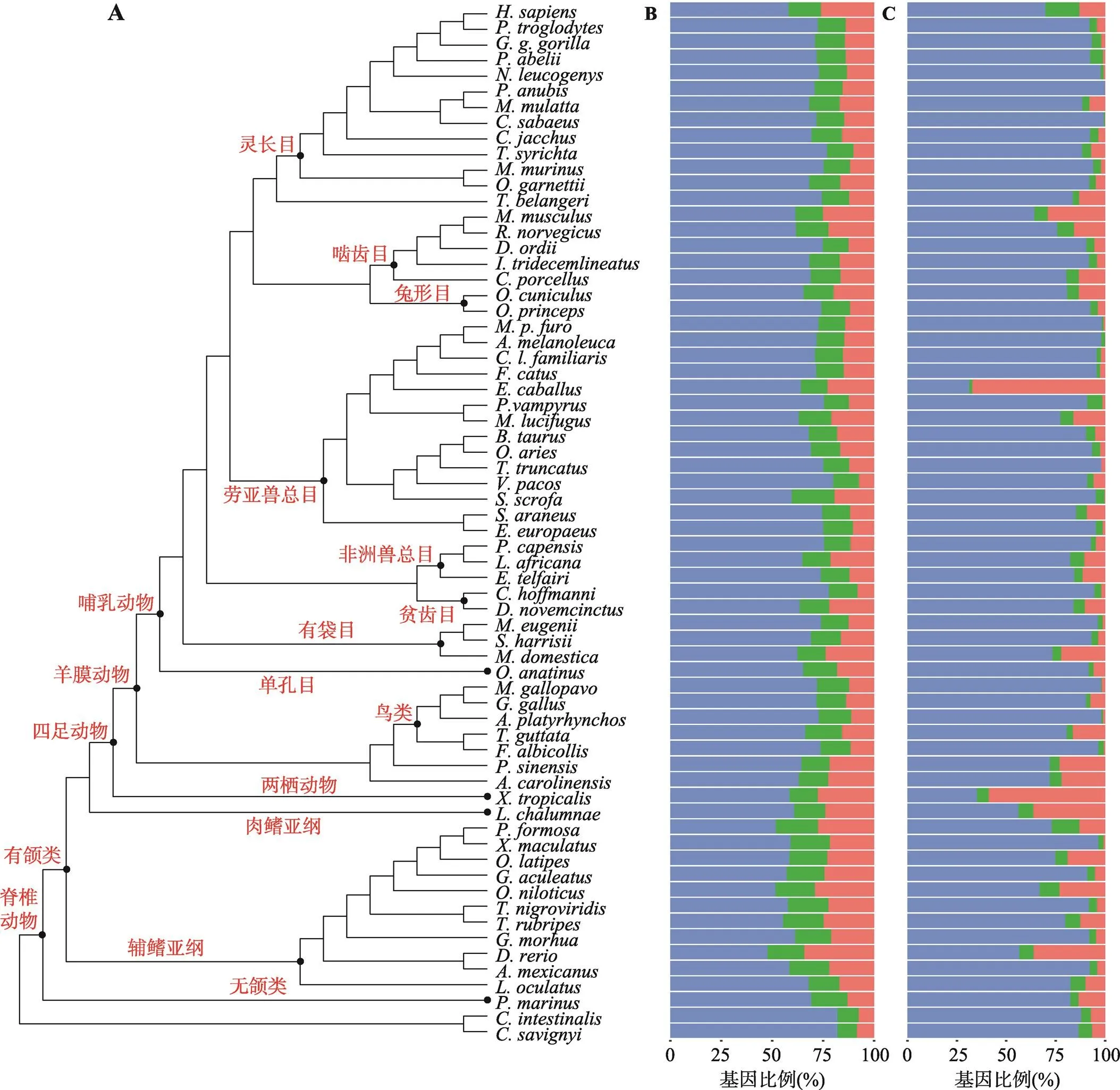
图1 脊椎动物系统发生关系及基因家族大小分布
A:66个物种的系统发育树(数据来自Ensembl v.84 数据库,图中黑色节点及相应的红色文字表示物种分类);B:各物种全基因组水平的基因家族大小分布;C:各物种Orphan基因家族大小分布(条形图中的蓝、绿、红分别表示单拷贝、双拷贝及多拷贝基因)。
2.3 Orphan基因家族的大小分布、特征及起源进化
2.3.1 Orphan基因家族大小的跨物种分布模式
特定物种基因组中的基因指的是在其他物种基因组中找不到其同源基因的一类基因[38],它们被认为与相应物种具有的特异的发育模式,适应特定的环境紧密相关[39]。本研究统计了上述66个物种基因组中各物种特异的基因家族及其成员基因的数量。结果表明,Orphan基因家族的数目和成员基因总数在这些物种中变异很大,如宽吻海豚基因组中仅有223个Orphan基因家族和相应的226个基因;而玻璃海鞘则具有最多的4956个Orphan基因家族,共包含5383个成员基因。各物种基因组中基因所占比例从1.4% (宽吻海豚)到19.4% (鸭嘴兽)不等(表1,表2)。
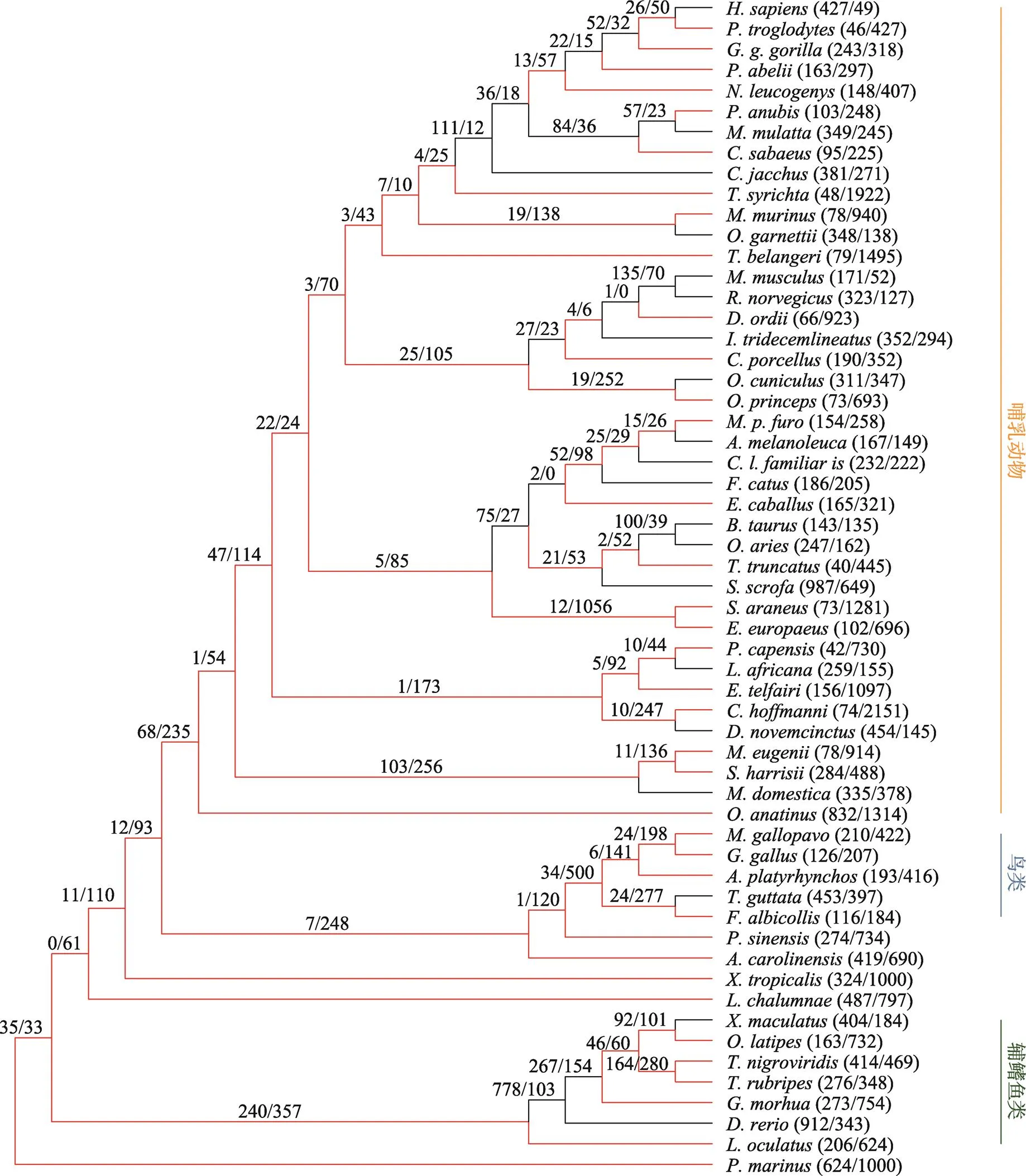
图2 脊椎动物中基因家族的扩增和收缩
分支上“/”线左右两侧的数字分别表示该分支上发生扩增及收缩的基因家族的数量;物种名称之后的数字表示相应物种基因组中发生扩增及收缩的基因家族的数量。黑色和红色分支分别表示从整体来说基因家族在特定分支上是扩增或收缩的。右侧橘色、蓝色和绿色的竖线分别标注了哺乳动物、鸟类及辐鳍鱼类在系统发育树中的位置。
进一步统计显示,基因在绝大部分物种中主要以单拷贝的形式存在,而斑马鱼、腔棘鱼、热带爪蟾和马中有相对较多的多拷贝的基因(图1C)。例如,马基因组有最高比例的多拷贝基因,这些基因分布在28个多拷贝的Orphan基因家族中,包含1055个基因,占该物种所有基因的67%。其中有两个家族分别含有514和271个基因,GO功能注释信息显示这些基因富集于RNA介导的转座这一功能类别。也就是说,马中多拷贝基因的高比例很可能是由逆转录转座产生了个别较大的基因家族而导致的。
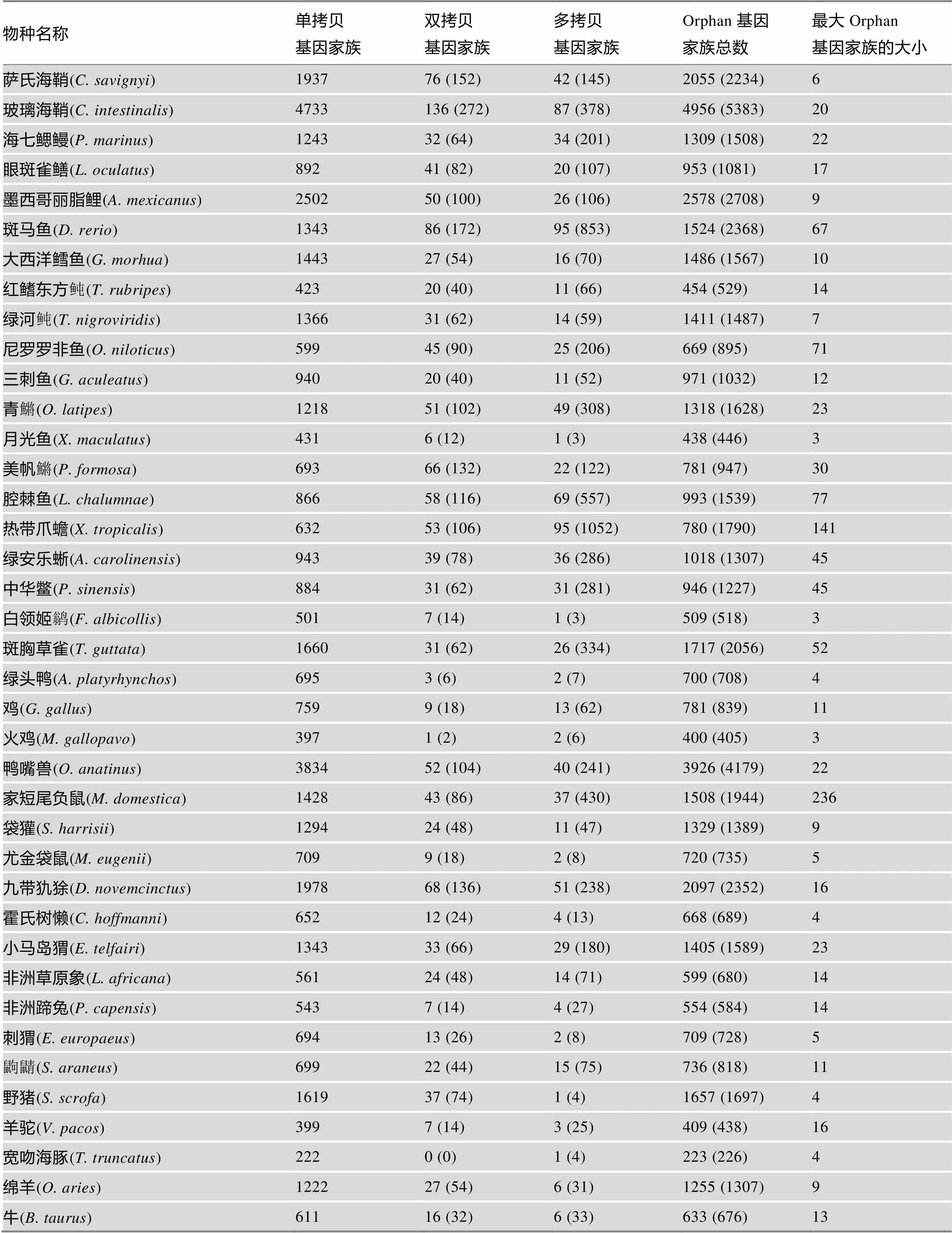
表2 物种特异性Orphan基因家族及成员基因的数量
续表
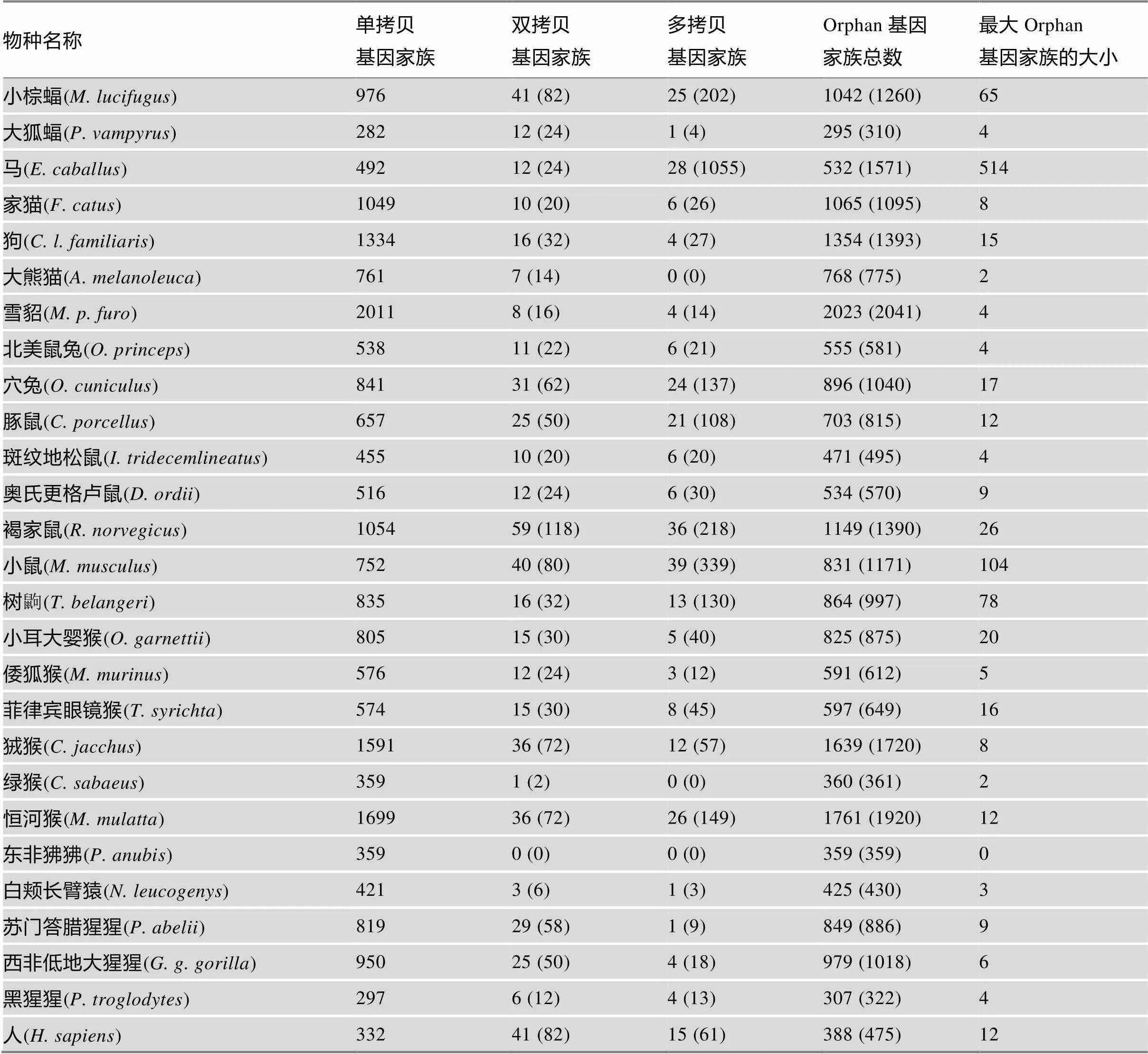
物种名称单拷贝基因家族双拷贝基因家族多拷贝基因家族Orphan基因家族总数最大Orphan基因家族的大小 小棕蝠(M. lucifugus)97641 (82)25 (202)1042 (1260)65 大狐蝠(P. vampyrus)28212 (24)1 (4)295 (310)4 马(E. caballus)49212 (24)28 (1055)532 (1571)514 家猫(F. catus)104910 (20)6 (26)1065 (1095)8 狗(C. l. familiaris)133416 (32)4 (27)1354 (1393)15 大熊猫(A. melanoleuca)7617 (14)0 (0)768 (775)2 雪貂(M. p. furo)20118 (16)4 (14)2023 (2041)4 北美鼠兔(O. princeps)53811 (22)6 (21)555 (581)4 穴兔(O. cuniculus)84131 (62)24 (137)896 (1040)17 豚鼠(C. porcellus)65725 (50)21 (108)703 (815)12 斑纹地松鼠(I. tridecemlineatus)45510 (20)6 (20)471 (495)4 奥氏更格卢鼠(D. ordii)51612 (24)6 (30)534 (570)9 褐家鼠(R. norvegicus)105459 (118)36 (218)1149 (1390)26 小鼠(M. musculus)75240 (80)39 (339)831 (1171)104 树鼩(T. belangeri)83516 (32)13 (130)864 (997)78 小耳大婴猴(O. garnettii)80515 (30)5 (40)825 (875)20 倭狐猴(M. murinus)57612 (24)3 (12)591 (612)5 菲律宾眼镜猴(T. syrichta)57415 (30)8 (45)597 (649)16 狨猴(C. jacchus)159136 (72)12 (57)1639 (1720)8 绿猴(C. sabaeus)3591 (2)0 (0)360 (361)2 恒河猴(M. mulatta)169936 (72)26 (149)1761 (1920)12 东非狒狒(P. anubis)3590 (0)0 (0)359 (359)0 白颊长臂猿(N. leucogenys)4213 (6)1 (3)425 (430)3 苏门答腊猩猩(P. abelii)81929 (58)1 (9)849 (886)9 西非低地大猩猩(G. g. gorilla)95025 (50)4 (18)979 (1018)6 黑猩猩(P. troglodytes)2976 (12)4 (13)307 (322)4 人(H. sapiens)33241 (82)15 (61)388 (475)12
括号中的数字表示成员基因的数量;基因家族的大小指基因家族中包含的基因个数。
2.3.2基因特征
以人类基因组中鉴定到的475个基因为例,分别从序列属性、表达水平、基因表达的组织特异性、功能注释等方面探究了基因的部分特征。
由图3A可知,基因编码的蛋白其序列长度显著低于非基因编码的蛋白(曼-惠特尼U检验,<2.20×10–16)。对该基因在16种人类组织的表达水平进行分析,发现这475个基因中只有292个基因有可利用的表达谱数据。与非基因相比,这些基因的表达水平较低(曼-惠特尼U检验,<2.20×10–16) (图3B);约60%的基因都是组织特异性表达(图3C),且主要倾向于在淋巴结中特异性表达(图3D),这暗示基因可能与免疫响应密切相关。
为了揭示出这些基因可能的生物学功能,本研究对基因进行功能富集分析。结果表明,尽管基因与非基因相比具有更高比例的未知功能基因(图4A),但已知功能的基因主要参与角质化、皮肤发育、上皮细胞分化、免疫响应等生物学过程(图4B)。
2.3.3 种系特异性基因家族的起源和进化
上述分析只涉及单个物种的特异性基因,而种系特异性基因对于理解特定分类学阶元的物种的基因组和表型进化也具有重要的意义。因此本研究进一步对种系特异性基因家族进行了鉴定。参考文献[40]中的方法,对于系统发育树上感兴趣的内部节点,当某基因家族包含了该节点下半数以上物种的基因时,该基因家族即被认为是相应节点起源的种系特异性基因家族。按照此原则,共有9488个种系特异性基因家族分布到脊椎动物主要类群系统发育树的节点上(图5)。
数据显示,从64个脊椎动物物种共同祖先起源的基因家族有1854个,脊椎动物在进化过程中,自有颌纲祖先物种起源的基因家族数量最多,为3839个。
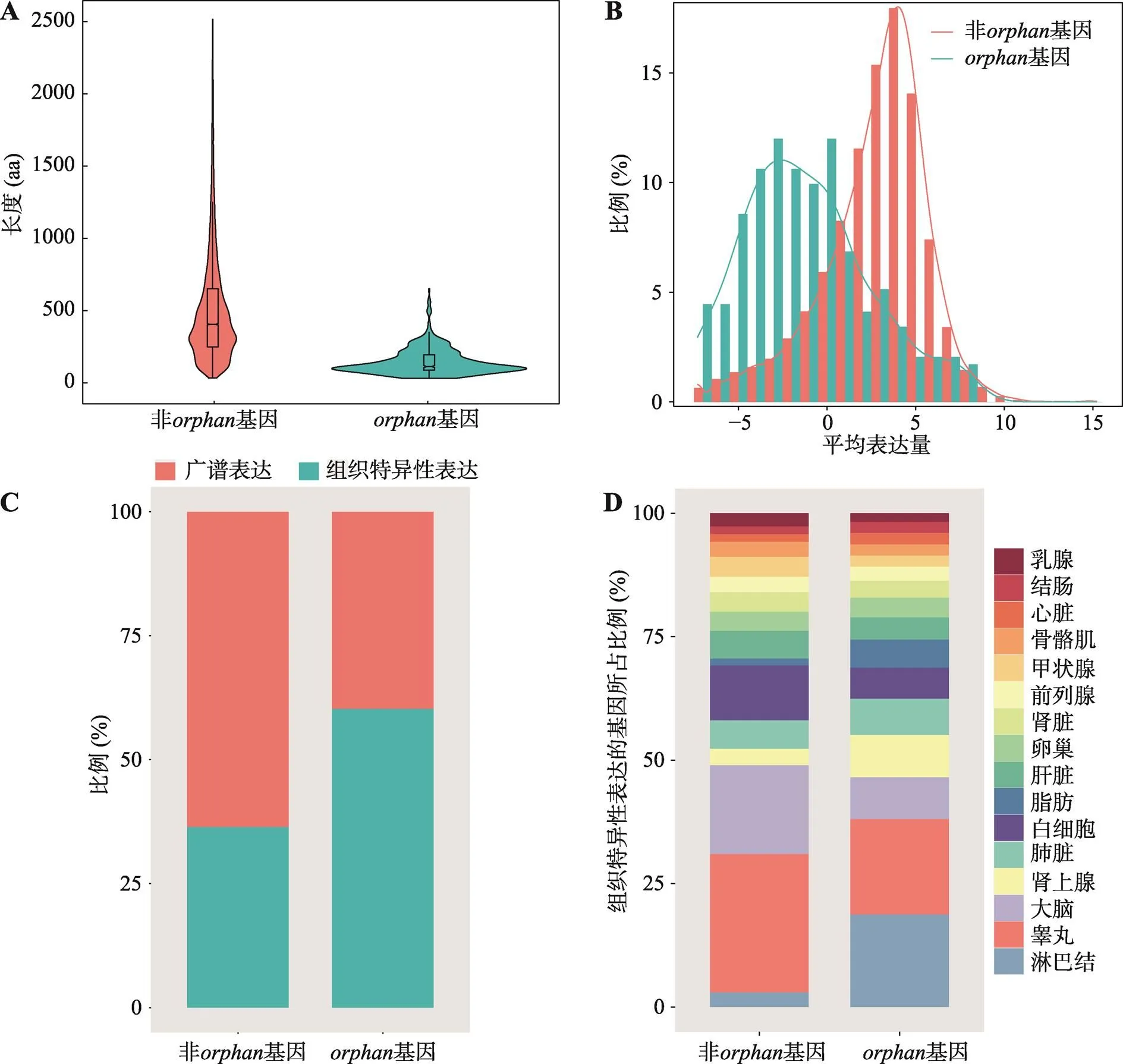
图3 orphan基因的序列长度与表达模式
A:人类基因组中基因、非基因编码的氨基酸序列长度;B:基因及非基因的表达水平(该图反映了特定表达水平(x轴)对应的基因所占的比例(y轴),每个基因的表达水平以所有样本中该基因表达水平的平均值取log来表示);C:基因与非基因中广谱表达基因及组织特异性表达基因所占的比例;D:组织特异性表达的基因和非基因在各组织中的分布。
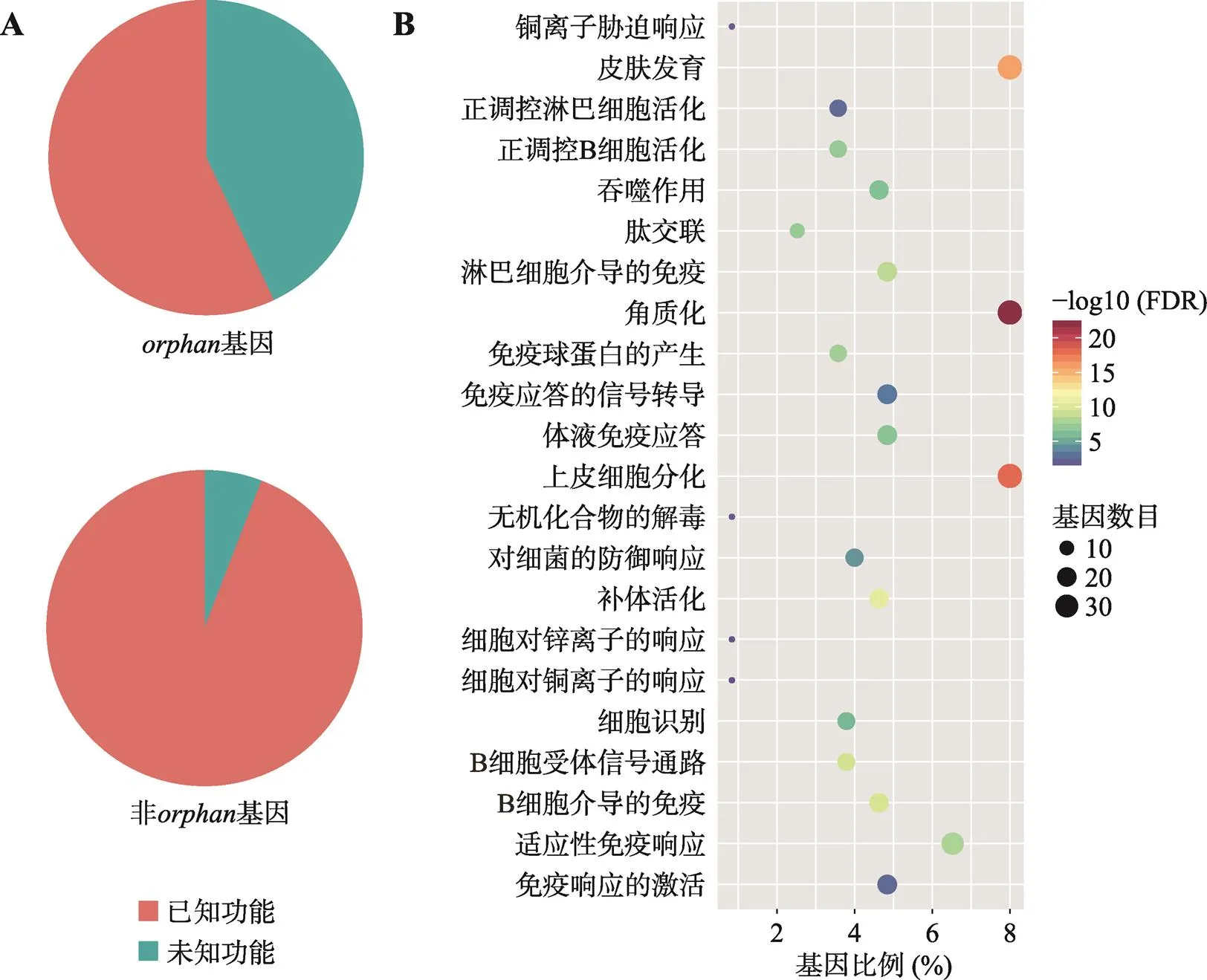
图4 orphan基因的功能注释
A:基因和非基因中有GO注释的基因所占的比例;B:基因的功能富集。
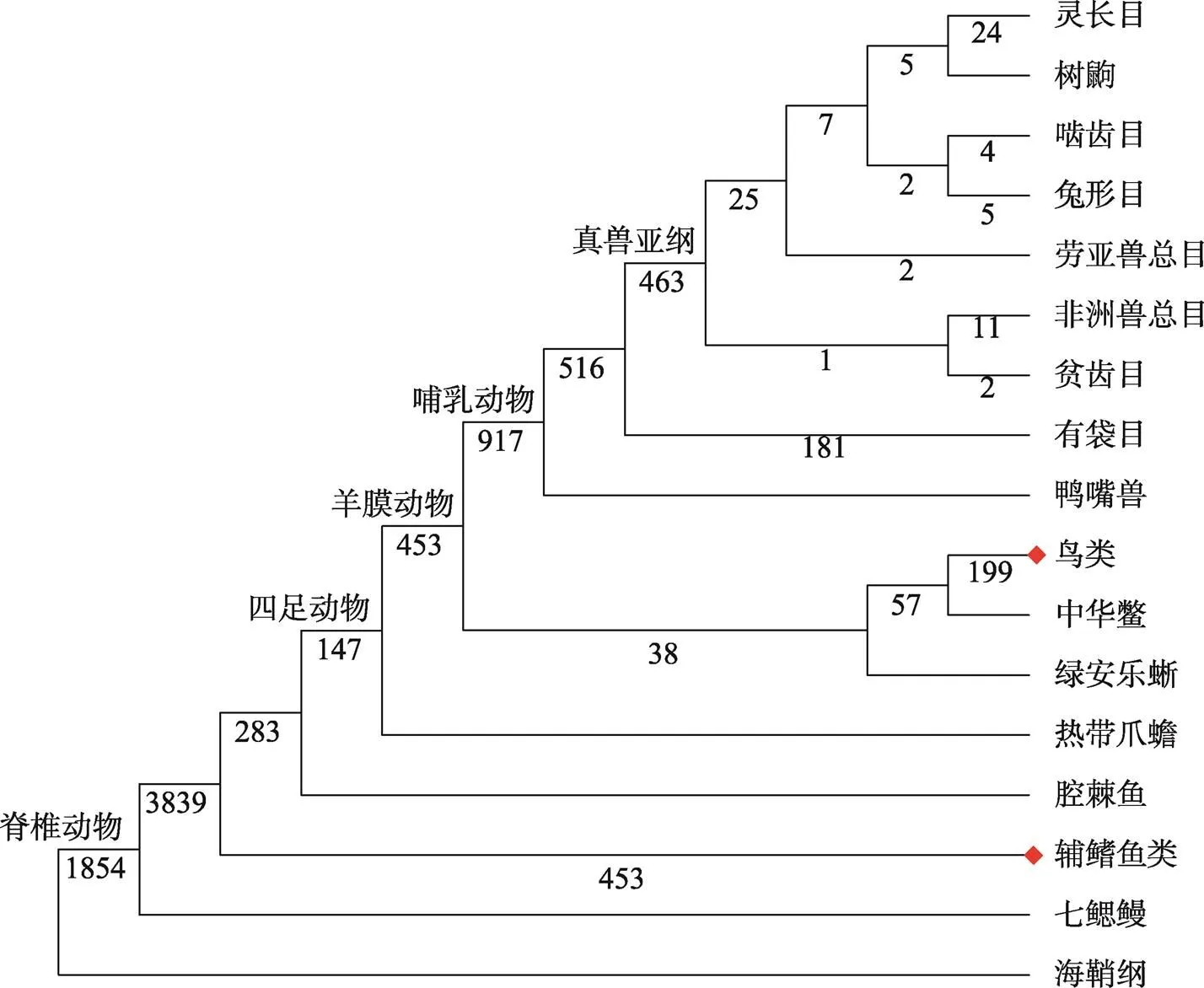
图5 脊椎动物不同种系中基因家族的数量

鱼类在早期进化中发生了一次该类群特异性的全基因组复制事件。数据显示辐鳍鱼特有的基因家族高达453个,推测这可能与鱼类特异的全基因组复制事件有关(图5)。当把腔棘鱼考虑在内时,硬骨鱼特有的基因家族则有183个,根据简约法原理,这些基因家族很可能是四足动物进化早期丢失的基因家族。为了调查这些基因是否对脊椎动物由水生到陆生的进化方式有贡献,本研究利用ZFIN (The Zebrafish Information Network)数据库中的基因表达、基因敲除、基因敲低数据对这些基因进行了分析。结果发现有84个基因其功能与鱼类特有的发育过程关系密切:分别有9个基因与鳍的发育有关,11个基因与躯干、体节、尾巴发育有关,7个基因与耳石及耳朵的发育有关,15个基因与肾脏发育有关,24个基因与眼睛及27个基因与大脑发育相关。这暗示脊椎动物从水生到陆生转变中某些关键特征的形成,如鳍到肢的转变、耳的重塑以及排氮形式的改变等与四足动物中特定基因的缺失有着密切联系。Amemiya等[41]鉴定到的55个在四足动物早期进化中丢失的基因中,有20个基因在本研究分析中得到了进一步证实。
为了探究硬骨鱼特有基因的表达特征,本研究以斑马鱼中的硬骨鱼特有基因为对象进行了分析。结果表明,斑马鱼中硬骨鱼特有的基因通常比非特有基因的表达水平低(图6A,曼-惠特尼U检验,< 2.20×10–16),但这些基因表达的组织特异性较高,且主要集中在鳃中特异性表达(图6B),这反映了硬骨鱼特有基因在硬骨鱼特异的发育过程中发挥了至关重要的作用。
本研究还调查了鸟类特有的基因家族。在199个鸟类特有的基因家族中,有134个家族含有共151个鸡的直系同源基因,GO功能富集分析(<0.05)显示其中许多基因参与了对细菌的防御响应以及与细胞骨架的结构成分有关。这些基因中有7个注释为羽毛角蛋白基因,分别是、、、、、和。与鱼类中观察结果相似,鸟类特有的基因相对非鸟类特有的基因通常表达水平更低(图6C,曼-惠特尼U检验,= 1.65×10–8)、表达的组织特异性更高,但无显著的组织偏好性(图6D)。
3 讨论
本研究对跨约6亿年进化时间的64个脊椎动物物种及2个海鞘纲外群物种进行了基因家族的鉴定和初步分析,揭示了脊椎动物基因家族大小的动态进化,并对部分基因家族拷贝数变异与特定分类群的宏进化之间的联系进行了推测。从全基因组水平来看,脊椎动物中的基因主要以单拷贝的形式存在,这与植物中观察到的现象不同。植物基因组中的基因大都以多基因家族的形式存在[20],这主要是由于植物中除了小规模复制外,还发生了非常广泛的全基因组复制事件。而脊椎动物中除了进化早期发生的两轮全基因组复制及真骨鱼类中额外的全基因组复制外,只在两栖类和辐鳍鱼部分物种中发现独立的全基因组复制事件[42,43]。
Demuth等[22]对人、黑猩猩、小鼠、大鼠和狗基因组中基因家族的扩增与收缩的研究发现,在灵长类动物中,人的基因组中有最少的基因丢失,而且相比之下,黑猩猩在相同时期内却丢失了更多基因。本研究也得到一致的结果,这在一定程度上揭示了这两个物种间表型差异背后的遗传变化。基因家族的大小受到各种因素的影响。基因复制、基因的起源等会增加基因家族的大小;而基因缺失(包括单个基因或染色体片段中几个基因的缺失)会使基因家族的大小减小[20]。除此之外,研究表明基因的功能也是决定基因家族大小的一个主要因素[22]。例如,脊椎动物中参与调控、信号转导、转录、蛋白质运输和蛋白质修饰的基因家族趋向于扩增,而参与新陈代谢过程的基因家族倾向于收缩。随机过程与自然选择是基因家族大小进化的驱动力[22]。有研究表明不同真核生物中基因家族的大小与选择压力的关系有所差异,如在单细胞真核生物酵母中,选择约束与基因家族的大小有很强的正相关关系,然而在多细胞真核生物中则呈现出弱的负相关[44]。
物种或种系特异性基因与其他物种中的基因序列不具有同源性,常被认为可能对物种的适应性进化有重要贡献[45~47]。本研究发现脊椎动物中的基因只在极个别物种中有较多的多拷贝,绝大多数仍以单拷贝的形式存在。与全基因组水平的基因家族大小分布相比,基因中单拷贝基因所占的比例高于全基因组中单拷贝基因的比例。这可能是由于脊椎动物中基因的复制能力低,或者是这些基因太“年轻”而没有足够的时间进化出额外的拷贝。此外,基因的产生机制比较特殊,该基因的形成贯穿整个进化历程并且是一个持续不断的过程,它不但可以通过复制和重排过程产生,也可以从基因组中的非编码区起源[48]。基因表达数据与功能注释等的结合进一步揭示了脊椎动物中物种或种系特异性基因的一般属性及其对物种适应性的影响。详细而言,这类基因通常编码的蛋白质序列长度较短、表达水平低、而表达的组织特异性高;硬骨鱼特有的基因中包含了对鱼类适应水生环境的重要基因,鸟类特异性基因中富集了羽毛角蛋白基因,这些分析证实了该类基因对物种或种系特异性表型创新的贡献。其中,对鸟类特异性基因的研究中,增加鸟类样本大小可能更有利于评估这类基因对鸟类特异性适应的影响。
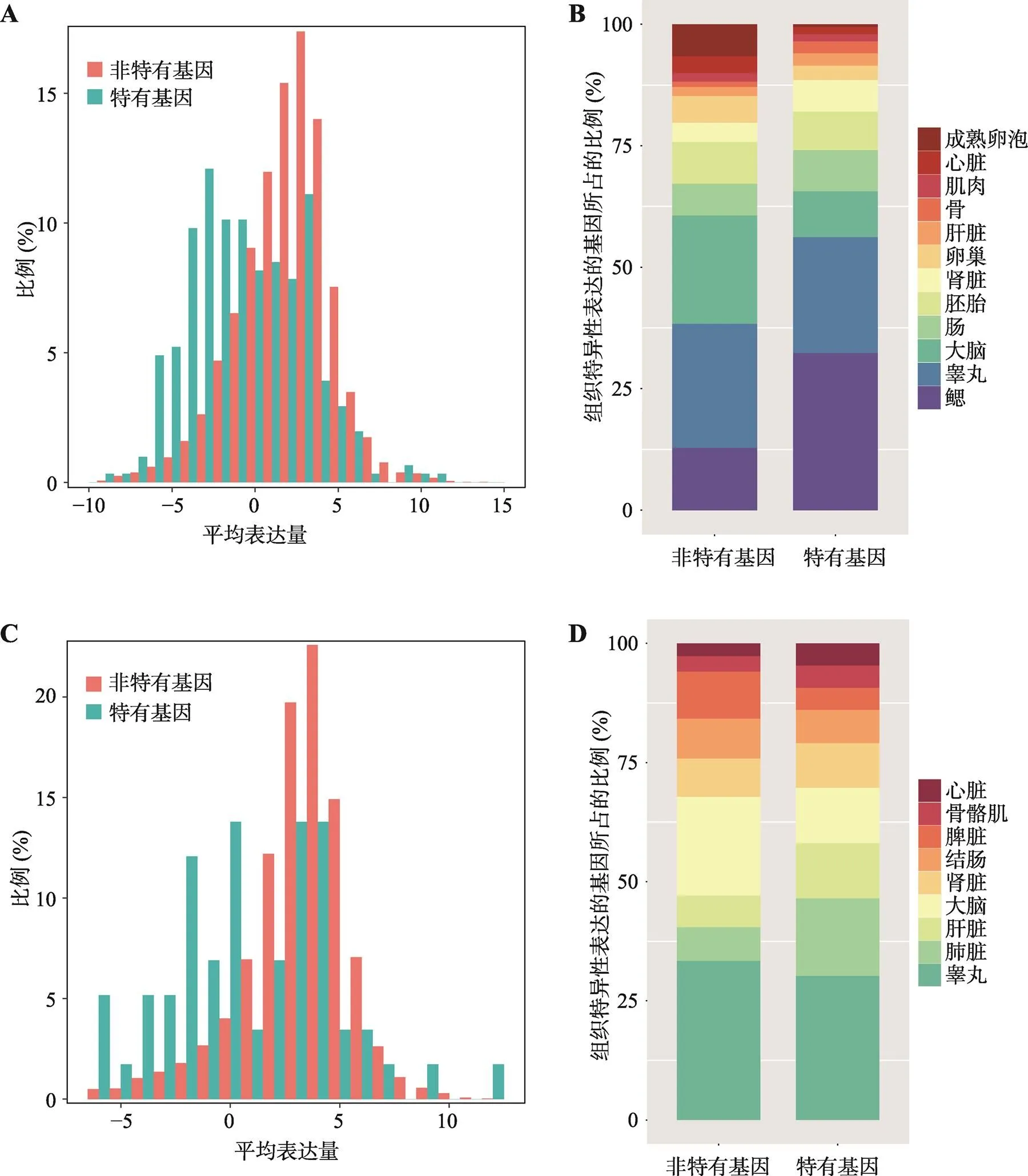
图6 硬骨鱼、鸟类特有基因的表达分析
A:斑马鱼基因组中所包含的硬骨鱼特有的基因及非特有基因的表达水平;B:组织特异性表达的硬骨鱼特有基因和非特有基因在斑马鱼各组织中的分布;C:鸡基因组中所含有的鸟类特有基因及非特有基因的表达水平;D:组织特异性表达的鸟类特有基因和非特有基因在鸡不同组织中的分布。
综上所述,本研究系统地阐述了脊椎动物进化过程中动态的基因得失过程导致的不同种系间基因家族大小的差异及其蕴含的生物学意义,对物种或种系特异的基因的分析为理解脊椎动物间表型的多样性提供了理论基础。
[1] Huang S, Chen Z, Yan X, Yu T, Huang G, Yan Q, Pontarotti PA, Zhao H, Li J, Yang P, Wang R, Li R, Tao X, Deng T, Wang Y, Li G, Zhang Q, Zhou S, You L, Yuan S, Fu Y, Wu F, Dong M, Chen S, Xu A. Decelerated genome evolution in modern vertebrates revealed by analysis of multiple lancelet genomes, 2014, 5: 5896.
[2] Blomme T, Vandepoele K, de Bodt S, Simillion C, Maere S, van de Peer Y. The gain and loss of genes during 600 million years of vertebrate evolution, 2006, 7(5): R43.
[3] Bosch N, Cáceres M, Cardone MF, Carreras A, Ballana E, Rocchi M, Armengol L, Estivill X. Characterization and evolution of the novel gene family FAM90A in primates originated by multiple duplication and rearrangement events, 2007, 16(21): 2572–2582.
[4] Zhang J. Evolution by gene duplication: an update, 2003, 18(6): 292–298.
[5] Peng GZ, Chen LL, Tian DC. Progress in the study of gene duplication., 2006, 28(7): 886–892.彭贵子, 陈玲玲, 田大成. 基因重复研究进展遗传, 2006, 28(7): 886–892.
[6] Albalat R, Cañestro C. Evolution by gene loss, 2016, 17(7): 379–391.
[7] Lin Q, Fan S, Zhang Y, Xu M, Zhang H, Yang Y, Lee AP, Woltering JM, Ravi V, Gunter HM, Luo W, Gao Z, Lim ZW, Qin G, Schneider RF, Wang X, Xiong P, Li G, Wang K, Min J, Zhang C, Qiu Y, Bai J, He W, Bian C, Zhang X, Shan D, Qu H, Sun Y, Gao Q, Huang L, Shi Q, Meyer A, Venkatesh B. The seahorse genome and the evolution of its specialized morphology, 2016, 540(7633): 395–399.
[8] Lespinet O, Wolf YI, Koonin EV, Aravind L. The role of lineage-specific gene family expansion in the evolution of eukaryotes, 2002, 12(7): 1048–1059.
[9] Li WH, Gu Z, Wang H, Nekrutenko A. Evolutionary analyses of the human genome, 2001, 409(6822): 847–849.
[10] Gu Z, Cavalcanti A, Chen FC, Bouman P, Li WH. Extent of gene duplication in the genomes of drosophila, nematode, and yeast, 2002, 19(3): 256– 262.
[11] Gilad Y, Man O, Glusman G. A comparison of the human and chimpanzee olfactory receptor gene repertoires, 2005, 15(2): 224–230.
[12] Johnson ME, Viggiano L, Bailey JA, Abdul-Rauf M, Goodwin G, Rocchi M, Eichler EE. Positive selection of a gene family during the emergence of humans and African apes, 2001, 413(6855): 514–519.
[13] McLysaght A, Baldi PF, Gaut BS. Extensive gene gain associated with adaptive evolution of poxviruses, 2003, 100(26): 15655–15660.
[14] Cheng CH, Chen L, Near TJ, Jin Y. Functional antifreeze glycoprotein genes in temperate-water New Zealand nototheniid fish infer an Antarctic evolutionary origin, 2003, 20(11): 1897–1908.
[15] Chen Z, Cheng CH, Zhang J, Cao L, Chen L, Zhou L, Jin Y, Ye H, Deng C, Dai Z, Xu Q, Hu P, Sun S, Shen Y, Chen L. Transcriptomic and genomic evolution under constant cold in antarctic notothenioid fish, 2008, 105(35): 12944–12949.
[16] Wang X, Grus WE, Zhang J. Gene losses during human origins, 2006, 4(3): e52.
[17] Fortna A, Kim Y, MacLaren E, Marshall K, Hahn G, Meltesen L, Brenton M, Hink R, Burgers S, Hernandez- Boussard T, Karimpour-Fard A, Glueck D, McGavran L, Berry R, Pollack J, Sikela JM. Lineage-specific gene duplication and loss in human and great ape evolution, 2004, 2(7): E207.
[18] Yu L, Wang GD, Ruan J, Chen YB, Yang CP, Cao X, Wu H, Liu YH, Du ZL, Wang XP, Yang J, Cheng SC, Zhong L, Wang L, Wang X, Hu JY, Fang L, Bai B, Wang KL, Yuan N, Wu SF, Li BG, Zhang JG, Yang YQ, Zhang CL, Long YC, Li HS, Yang JY, Irwin DM, Ryder OA, Li Y, Wu CI, Zhang YP. Genomic analysis of snub-nosed monkeys () identifies genes and processes related to high-altitude adaptation, 2016, 48(8): 947–952.
[19]Genome Project. Thegenome and the evolution of flowering plants, 2013, 342 (6165): 1241089.
[20] Guo YL. Gene family evolution in green plants with emphasis on the origination and evolution ofthaliana genes, 2013, 73(6): 941–951.
[21] Demuth JP, de Bie T, Stajich JE, Cristianini N, Hahn MW. The evolution of mammalian gene families, 2006, 1: e85.
[22] Prachumwat A, Li WH. Gene number expansion and contraction in vertebrate genomes with respect to invertebrate genomes, 2008, 18(2): 221–232.
[23] Meadows JRS, Lindblad-Toh K. Dissecting evolution and disease using comparative vertebrate genomics, 2017, 18(10): 624–636.
[24] Li L, Stoeckert CJJr., Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes, 2003, 13(9): 2178–2189.
[25] Enright AJ, van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families, 2002, 30(7): 1575–1584.
[26] de Bie T, Cristianini N, Demuth JP, Hahn MW. CAFE: a computational tool for the study of gene family evolution, 2006, 22(10): 1269–1271.
[27] Hedges SB, Dudley J, Kumar S. TimeTree: A public knowledge-base of divergence times among organisms, 2006, 22(23): 2971–2972.
[28] Hahn MW, de Bie T, Stajich JE, Nguyen C, Cristianini N. Estimating the tempo and mode of gene family evolution from comparative genomic data, 2005, 15(8): 1153–1160.
[29] Hahn MW, Han MV, Han SG. Gene family evolution across 12genomes, 2007, 3(11): e197.
[30] Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, Lancet D, Shmueli O. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification, 2005, 21(5): 650–659.
[31] Ruiz-Orera J, Hernandez-Rodriguez J, Chiva C, Sabidó E, Kondova I, Bontrop R, Marqués-Bonet T, Albà MM. Origins ofgenes in human and chimpanzee, 2015, 11(12): e1005721.
[32] Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, Storz JF, Antunes A, Greenwold MJ, Meredith RW, Ödeen A, Cui J, Zhou Q, Xu L, Pan H, Wang Z, Jin L, Zhang P, Hu H, Yang W, Hu J, Xiao J, Yang Z, Liu Y, Xie Q, Yu H, Lian J, Wen P, Zhang F, Li H, Zeng Y, Xiong Z, Liu S, Zhou L, Huang Z, An N, Wang J, Zheng Q, Xiong Y, Wang G, Wang B, Wang J, Fan Y, da Fonseca RR, Alfaro-Núñez A, Schubert M, Orlando L, Mourier T, Howard JT, Ganapathy G, Pfenning A, Whitney O, Rivas MV, Hara E, Smith J, Farré M, Narayan J, Slavov G, Romanov MN, Borges R, Machado JP, Khan I, Springer MS, Gatesy J, Hoffmann FG, Opazo JC, Håstad O, Sawyer RH, Kim H, Kim KW, Kim HJ, Cho S, Li N, Huang Y, Bruford MW, Zhan X, Dixon A, Bertelsen MF, Derryberry E, Warren W, Wilson RK, Li S, Ray DA, Green RE, O'Brien SJ, Griffin D, Johnson WE, Haussler D, Ryder OA, Willerslev E, Graves GR, Alström P, Fjeldså J, Mindell DP, Edwards SV, Braun EL, Rahbek C, Burt DW, Houde P, Zhang Y, Yang H, Wang J, Avian GC, Jarvis ED, Gilbert MT, Wang J. Comparative genomics reveals insights into avian genome evolution and adaptation, 2014, 346(6215): 1311– 1320.
[33] Kapusta A, Suh A, Feschotte C. Dynamics of genome size evolution in birds and mammals, 2017, 114(8): E1460–E1469.
[34] Hoegg S, Brinkmann H, Taylor JS, Meyer A. Phylogenetic timing of the fish-specific genome duplication correlates with the diversification of teleost fish, 2004, 59(2): 190–203.
[35] van de Peer Y, Maere S, Meyer A. The evolutionary significance of ancient genome duplications, 2009, 10(10): 725–732.
[36] Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer, 2005, 5(8): 615–625.
[37] Zhang Q, Su B. Evolutionary origin and human-specific expansion of a cancer/testis antigen gene family, 2014, 31(9): 2365–2375.
[38] Fischer D, Eisenberg D. Finding families for genomic ORFans, 1999, 15(9): 759–762.
[39] Long M, Betrán E, Thornton K, Wang W. The origin of new genes: glimpses from the young and old, 2003, 4(11): 865–875.
[40] Luis Villanueva-Cañas J, Ruiz-Orera J, Agea MI, Gallo M, Andreu D, Albà MM. New genes and functional innovation in mammals, 2017, 9(7): 1886–1900.
[41] Amemiya CT, Alföldi J, Lee AP, Fan S, Philippe H, Maccallum I, Braasch I, Manousaki T, Schneider I, Rohner N, Organ C, Chalopin D, Smith JJ, Robinson M, Dorrington RA, Gerdol M, Aken B, Biscotti MA, Barucca M, Baurain D, Berlin AM, Blatch GL, Buonocore F, Burmester T, Campbell MS, Canapa A, Cannon JP, Christoffels A, de Moro G, Edkins AL, Fan L, Fausto AM, Feiner N, Forconi M, Gamieldien J, Gnerre S, Gnirke A, Goldstone JV, Haerty W, Hahn ME, Hesse U, Hoffmann S, Johnson J, Karchner SI, Kuraku S, Lara M, Levin JZ, Litman GW, Mauceli E, Miyake T, Mueller MG, Nelson DR, Nitsche A, Olmo E, Ota T, Pallavicini A, Panji S, Picone B, Ponting CP, Prohaska SJ, Przybylski D, Saha NR, Ravi V, Ribeiro FJ, Sauka-Spengler T, Scapigliati G, Searle SM, Sharpe T, Simakov O, Stadler PF, Stegeman JJ, Sumiyama K, Tabbaa D, Tafer H, Turner-Maier J, van Heusden P, White S, Williams L, Yandell M, Brinkmann H, Volff JN, Tabin CJ, Shubin N, Schartl M, Jaffe DB, Postlethwait JH, Venkatesh B, Di Palma F, Lander ES, Meyer A, Lindblad-Toh K. The African coelacanth genome provides insights into tetrapod evolution, 2013, 496(7445): 311–316.
[42] Mable BK, Alexandrou MA, Taylor MI. Genome duplication in amphibians and fish: an extended synthesis, 2011, 284(3): 151–182.
[43] Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S, Fukui A, Hikosaka A, Suzuki A, Kondo M, van Heeringen SJ, Quigley I, Heinz S, Ogino H, Ochi H, Hellsten U, Lyons JB, Simakov O, Putnam N, Stites J, Kuroki Y, Tanaka T, Michiue T, Watanabe M, Bogdanovic O, Lister R, Georgiou G, Paranjpe SS, van Kruijsbergen I, Shu S, Carlson J, Kinoshita T, Ohta Y, Mawaribuchi S, Jenkins J, Grimwood J, Schmutz J, Mitros T, Mozaffari SV, Suzuki Y, Haramoto Y, Yamamoto TS, Takagi C, Heald R, Miller K, Haudenschild C, Kitzman J, Nakayama T, Izutsu Y, Robert J, Fortriede J, Burns K, Lotay V, Karimi K, Yasuoka Y, Dichmann DS, Flajnik MF, Houston DW, Shendure J, DuPasquier L, Vize PD, Zorn AM, Ito M, Marcotte EM, Wallingford JB, Ito Y, Asashima M, Ueno N, Matsuda Y, Veenstra GJ, Fujiyama A, Harland RM, Taira M, Rokhsar DS. Genome evolution in the allotetraploid frog, 2016, 538(7625): 336–343.
[44] Conant GC, Wagner A. Genomehistory: a software tool and its application to fully sequenced genomes, 2002, 30(15): 3378–3386.
[45] Wilson GA, Bertrand N, Patel Y, Hughes JB, Feil EJ, Field D. Orphans as taxonomically restricted and ecologically important genes, 2005, 151(Pt8): 2499– 2501.
[46] Zhang YE, Long M. New genes contribute to genetic and phenotypic novelties in human evolution, 2014, 29: 90–96.
[47] Toll-Riera M, Bosch N, Bellora N, Castelo R, Armengol L, Estivill X, Albà MM. Origin of primate orphan genes: a comparative genomics approach, 2009, 26(3): 603–612.
[48] Tautz D, Domazet-Lošo T. The evolutionary origin of orphan genes, 2011, 12(10): 692–702.
Comparative analysis of gene family size provides insight into the adaptive evolution of vertebrates
Yu Meng, Ruolin Yang
Copy numbers of homologous gene families vary greatly among different species, which is caused by the differences in the rates of gene gain and loss. It is well known that gene copy number variation can be responsible for the phenotypic novelties of particular species. In this study, 64 species that represent the main vertebrate groups spanning evolutionary period of about 600 million years were selected and the homology of gene families across these species were established, thereby revealing the evolutionary patterns of gene family size in vertebrates. The results show that among the 6857 gene families inferred to be present in the most recent common ancestor of the vertebrates, 6712 had changed their sizes in at least one lineage, and these gene families had contracted in most cases. Gene families inandhad undergone the greatest contraction and expansion, respectively. Based on the highly dynamic evolution of vertebrate gene family size, we sought to identify any genomic signals that might be related to the evolution of specific vertebrate populations from the perspective of the distinct gene family size changes. We observed a high proportion of gene family amplification occurred, probably due to genome-wide duplication in the recent common ancestral genome of teleosts, which was followed by contraction in the decedents due to the extensive gene fractionation. Furthermore, we found evidence that orphan genes in the bony fish might contribute to the adaptive evolution of fish in aquatic environment. For example, some orphan genes were involved in fin development, tail development and kidney physiology. Overall, our work provides novel insights into the evolution of vertebrate gene family size and provides several lines of evidence for understanding the relationship between the genome evolution and phenotypic diversity in vertebrates.
vertebrates; gene family; adaptive evolution;gene
2018-08-06;
2018-12-13
孟玉,硕士研究生,专业方向:遗传学。E-mail: m1994yu@163.com
杨若林,教授,博士生导师,研究方向:进化遗传学和生物信息学。E-mail: desert.ruolin@gmail.com
10.16288/j.yczz.18-225
2019/1/14 13:15:21
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190114.1315.004.html
(责任编委: 于黎)
