基于转录组数据的网络分析挖掘鼻咽癌与口腔鳞癌的共享功能模块
2019-02-28陈应坚廖苑君林帆孙胜南赵小蕾覃继恒饶绍奇
陈应坚,廖苑君,林帆,孙胜南,赵小蕾,覃继恒,饶绍奇
基于转录组数据的网络分析挖掘鼻咽癌与口腔鳞癌的共享功能模块
陈应坚1,2,廖苑君1,2,林帆1,2,孙胜南1,2,赵小蕾2,覃继恒2,饶绍奇2
1. 广东医科大学公共卫生学院,东莞 523808 2.广东医科大学医学系统生物学研究所,东莞 523808
鼻咽癌和口腔鳞癌是两种在临床上高度相关的疾病,从分子层面系统性研究这两种疾病的相互关系却鲜见报道。本研究通过大规模的转录组数据分析识别鼻咽癌和口腔鳞癌的共享功能模块及其核心基因(一因多效模块和基因),以期阐明这两种疾病共享的分子机制。从GEO数据库获取这两种癌症的两套转录组数据,应用倍数法和经验贝叶斯方法筛选出鼻咽癌差异表达基因1279个,口腔鳞癌差异表达基因1293个,其中两者共享基因278个。以共享基因为种子,通过蛋白质-蛋白质互作知识引导构建基因网络,其中最大子网包含1290个基因和1766互作对。应用Newman算法提取了15个共享功能模块。对这些模块进行拓扑学分析,挖掘出58个核心基因,包括已知的与鼻咽癌或口腔鳞癌相关的基因(如、、、和等)和鲜有报道的基因(如、、、和等)。通路富集分析发现鼻咽癌和口腔鳞癌的共享功能模块参与多个生物学通路,包括p53信号通路、ECM受体相互作用、黏着斑、细胞周期等。本研究表明鼻咽癌和口腔鳞癌具有相似的致癌机制,所挖掘的共享模块可能是这两种疾病演化的核心分子相互作用机制。
鼻咽癌;口腔鳞癌;基因芯片;网络分析;基因多效性
鼻咽癌(nasopharyngeal carcinoma, NPC)是一种起源于鼻咽上皮的鳞状细胞癌[1],在中国南方有很高的发病率。口腔鳞状细胞癌(oral squamous cell carcinoma, OSCC)是发生于口腔黏膜上皮的恶性肿瘤,是全球性第六大常见癌症[2]。NPC和OSCC在组织类型、流行病学、发病机制及治疗方法上具有较高的相似性和相关性,提示两者之间拥有共同的致病因素,而这个致病因素则可能来自共享的遗传因子—基因。这种一个基因座(locus)影响两个或多个性状(phenotypic trait)的现象称为基因多效性[3](pleiotropy),在癌症的遗传机制中普遍存在。例如,抑癌基因在细胞中具有磷酸酶依赖性活性和非磷酸酶依赖性活性,并控制着多种生物过程,包括维持基因组的稳定性、细胞存活、迁移、增殖和代谢,是已知的与多种癌症相关的多效性基因[4]。因此,挖掘NPC与OSCC的共享风险功能模块及其基因对寻找更有效的诊断方法,深度研究两者发病机制以及有效地预防NPC和OSCC具有重要意义。
近年来,基因表达谱数据迅速增加,利用生物信息学方法对基因表达谱数据进行深入研究已成为一个新的研究热点。目前对单一疾病的基因生物信息学的研究较多,然而对多种疾病的基因生物信息学的研究较少,因此,本研究利用大规模的转录数据来寻找NPC和OSCC的共享风险基因及其共享基因功能模块,以探索这两种疾病共享的分子机制。
1 材料与方法
1.1 数据来源
从GEO数据库(Gene Expression Omnibus, http:// www.ncbi.nlm.nih.gov/geo/)中分别下载NPC和OSCC基因表达谱芯片原始数据。选取的NPC数据集编号是GSE12452,平台编号是GPL570。选取的OSCC数据集编号是GSE3524,平台编号是GPL96。用于网络构建的蛋白质-蛋白质互作(protein-protein interaction, PPI)数据来源为人类蛋白互作数据库(Human Protein Reference Database, HPRD)。目前HPRD数据库收录了41 327对蛋白互作数据(版本号为Release 9),是目前数据库中关于人类蛋白互作数据可信度最高的数据库之一。
1.2 芯片数据的预处理
预处理通过质量控制,剔除不及格的芯片数据,只保留及格的进入下一步处理。然后通过标准化,将芯片数据中的基因表达值变换到一个可以比较的水平。本研究采用NUSE箱线图完成数据的质量控制。若所有芯片的质量都非常可靠,则NUSE值都在1附近;若NUSE值大于1.05,则芯片质量就有问题。本研究下载的基因芯片原始数据统一用RMA (Robust Multi-array Analysis)一体化算法对数据进行标准化和对数化。该过程利用R语言“affy”软件包完成。
1.3 差异表达基因的筛选
采用倍数法(fold change, FC)和经验贝叶斯方法(empirical Bayes, EB)评估基因在病例对照中的差异表达程度和统计显著性。本研究采用相应实验平台的Affymetrix注释文件分别将两套芯片的探针编号注释为基因名称,若多个探针对应一个基因,则取所有探针的平均值作为基因的表达值。设定值小于0.01且logFC的绝对值大于1,即FC>2作为阈值分别筛选NPC和OSCC的差异表达基因,然后对两种疾病的差异表达基因取交集,得到二者的共享风险基因。该过程利用R语言“limma”软件包完成。
1.4 NPC与OSCC共享风险基因网络的构建
将筛选得到的NPC与OSCC共享风险基因作为初始种子基因(seed gene)。然后,将初始种子基因及其在PPI中的一级邻居基因,作为本研究构建NPC与OSCC共享风险基因网络的节点集合,对于集合中任意两个不同基因,如果其基因产物在PPI网络中存在互作,即视为网络中的连线,从而构建的NPC与OSCC共享风险基因网络,通过Cytoscape软件(版本3.6.1)进行PPI网络可视化。
1.5 识别NPC与OSCC共享风险功能模块
功能模块在蛋白互作网络中的一个重要特征是功能模块内的基因间存在高度紧密的相互作用和联系,而模块内连接密度相比与其他功能模块间的联系要高很多。基于该研究假设,本研究拟对所构建的NPC与OSCC共享风险基因网络进行模块的划分。本研究采用Newman算法[5,6]对NPC与OSCC共享风险基因网络进行网络分解,挖掘网络中高度模块化的功能模块。该算法将网络分解问题转变成二次型优化问题,即求使目标函数最大的列向量的取值:

其中是初始种子基因及其在PPI中的一级邻居基因组成的集合,是集合的邻接矩阵(adjacent matrix),用于表示网络中基因间的相互连接关系,A=1表示网络中基因和基因存在蛋白互作,反之A=0。是网络中边的数目,k和k分别代表节点和节点的连通度。是取值为1或-1的指示向量,s表示基因所属模块的情况,即
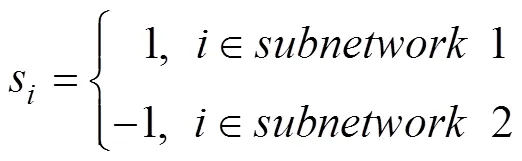
根据柯西(Cauchy)不等式,当取矩阵的最大特征值对应的特征向量时,目标函数达到最大值。由于只取1或-1,因此只保留特征向量对应元素的正负号,从而根据特征向量的正负号将网络分解为两个子模块,重复这个过程,直到网络不能继续分解时,即可完成对网络的分解。最终,本研究将节点数大于30的子网络定义为网络模块。
1.6 网络拓扑属性分析与核心基因的识别
本研究对挖掘出的每个功能模块的拓扑属性进行评价,评价指标包括:
(1)网络直径:指网络中任意两个连通节点间距离的最大值。网络直径代表了网络中任意两个节点间可能出现的最远距离,可从侧面反映网络的紧密性。
(2)特征路径长度:即模块中所有节点间的最短路径的平均值,反映了模块的紧密程度;
(3)连通度:即模块中直接与某一节点相连的边的数目,它不仅反映了单一节点在模块中的最基本的拓扑性质,而且是判断无标度网络属性的重要指标。无标度网络是网络中连通度的分布符合幂律分布,即()∝a,其中为接通度,α是指数参数,可通过极大似然法估计。通过Kolmogorov-Smirnov (KS)拟合优度检验评判网络的无标度性质。
无标度网络的属性很大程度上由少数的节点即核心基因(hub gene)决定的。虽然网络具有抗随机扰动的能力,但对中心节点的作用却很敏感。核心基因的识别一般通过检验其在网络中的连通度是否高出随机网络下的连通度期望值。该零假设可以通过泊松分布进行检验。那么,在一个节点数为N的随机网络中,某节点连通度等于的概率为:
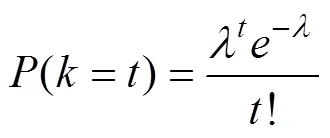
其中,=N,表示随机网络中节点的期望连通度,是网路中任意两个节点发生连接的概率。为控制假阳性率,FDR<0.01作为显著性水平。上述网络分析通过R软件实现,其中部分分析借助“igraph”软件包完成。
1.7 KEGG通路富集分析
KEGG (Kyoto Encyclopedia of Genes and Genomes)通路通过描述基因编码的生物大分子酶或者蛋白质间相互联系相互影响的情况以阐释基因及其产物的功能[7]。本研究利用在线注释网站DAVID软件(版本6.8)对每一个模块进行KEGG信号通路富集分析。为控制假阳性率,采用Fisher精确检验的方法,富集结果取FDR<0.001作为显著性基因富集。
2 结果与分析
2.1 芯片数据的预处理
从GEO数据库获取NPC与OSCC基因表达谱芯片原始数据后,对原始数据进行预处理。采用NUSE箱线图的数据处理结果分别见图1A和图1B。结果显示,NPC的正常样本GSM312899、GSM312902和OSCC的正常样本GSM80520、肿瘤样本GSM80471的NUSE 值均大于1.05,说明这4个样本的芯片存在质量问题。为保证后续分析计算结果的可信度,删除NPC的正常样本GSM312899、GSM312902和OSCC的正常样本GSM80520、肿瘤样本GSM80471,最终获得的有效样本包括NPC正常样本8个和肿瘤样本31个,OSCC正常样本3个和肿瘤样本15个。基于RMA一体化算法,使用R语言“affy”包对两种疾病的样本芯片数据分别进行标准化和对数化。
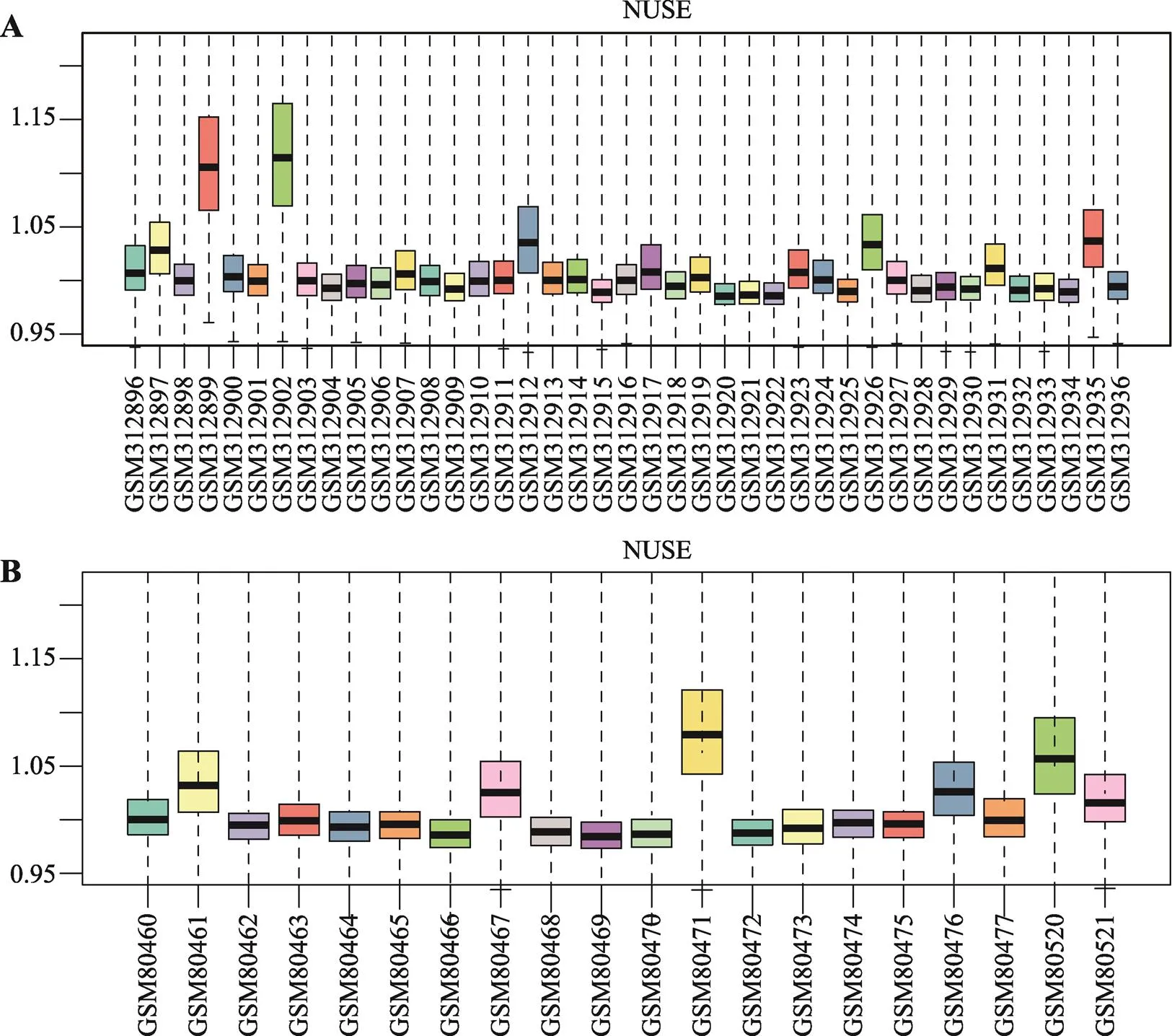
图1 NUSE箱线图
A:NPC基因表达谱芯片的NUSE箱线图;B:OSCC基因表达谱芯片的NUSE箱线图。
2.2 差异表达基因的筛选
采用倍数法和经验贝叶斯检验方法,按照值小于0.01且logFC的绝对值大于1为阈值,共筛选出NPC差异表达基因1279个,OSCC差异表达基因1293个,二者的共同差异表达基因278个。
2.3 NPC与OSCC共享风险基因网络的构建
通过提取种子基因及其在PPI网络中的一级邻居,最终构建出38个大小不同的独立的基因互作网络,包含1412个节点和1851条边,其中节点表示蛋白(或称基因),边表示蛋白互作,见图2。最大子网络由1290个节点和1766条边组成。后面的分析均基于网络中的最大子网进行。
2.4 疾病网络模块与核心基因的识别
利用Newman 算法对最大子网进行分解,最终得15个节点数大于30的功能模块。表1列出了各模块的基本特征和拓扑属性。可以发现规模大的模块其包含的初始种子基因相对多,平均每个节点连接的边数也多。不同网络节点数相差也很大,最大的模块(M1)由196个基因(其中初始种子基因有18个)及270条边,平均每个节点含有1.4条边,然而最小的模块(M14和M15)由34个基因(其中初始种子基因分别有3个和9个)和34条边组成,平均每个节点含有1条边。这些模块的拓扑属性接近,网络直径变化范围在4~9,特征路径长度均小于6,符合“六度分离理论”[8],各模块的指数参数α估计值在2~3之间,符合生物学上无标度网络的常见表现。本研究对节点连通度是否符合幂律分布进行KS拟合优度检验,检验结果显示各模块的连通度均近似服从幂律分布(>0.05),支持了各模块的无标度性,可认为是无标度网络。
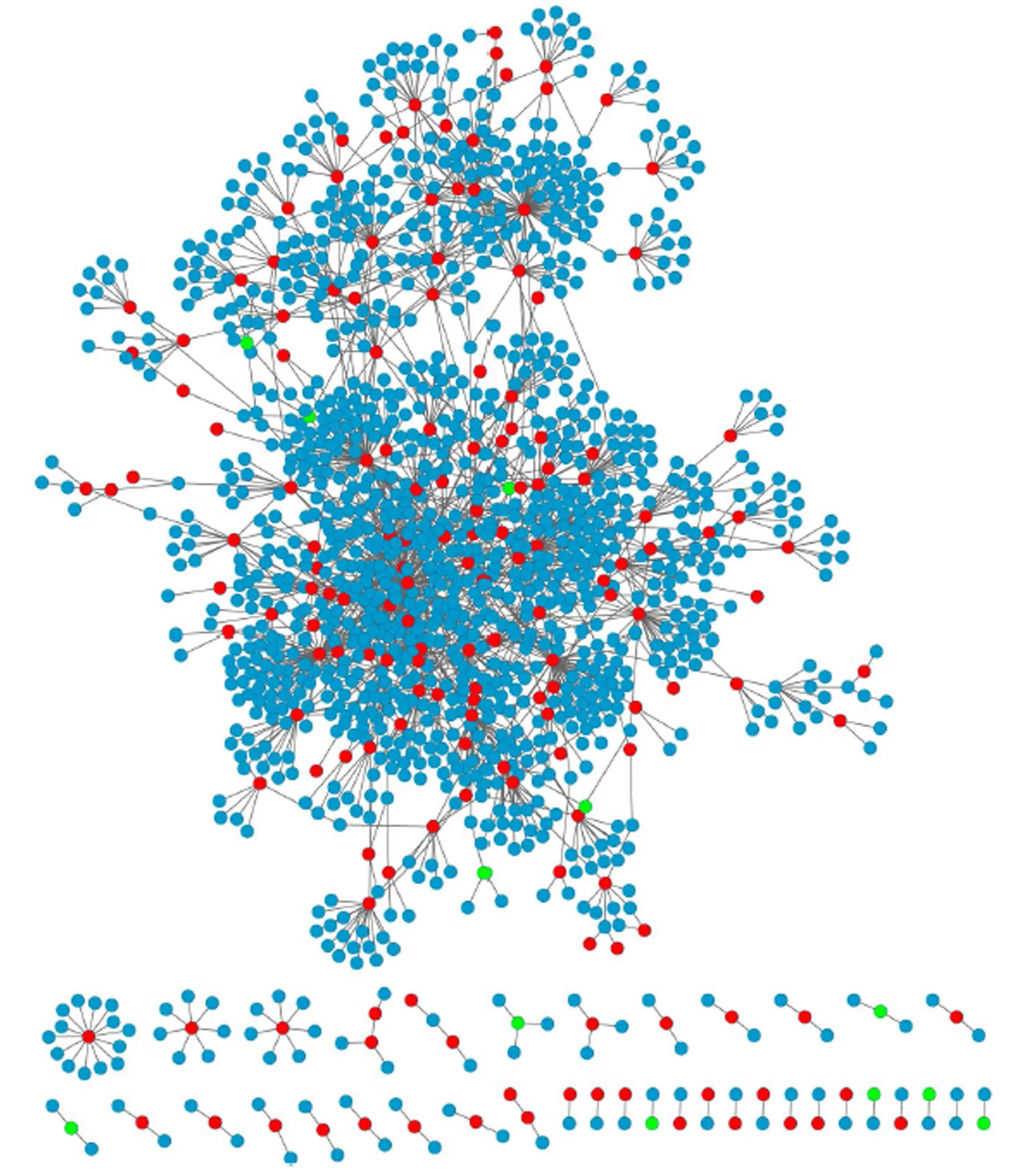
图2 NPC与OSCC共享的差异表达基因的PPI网络图
红色节点表示上调差异表达基因编码蛋白;绿色表示下调差异表达基因编码蛋白,蓝色表示其他蛋白。
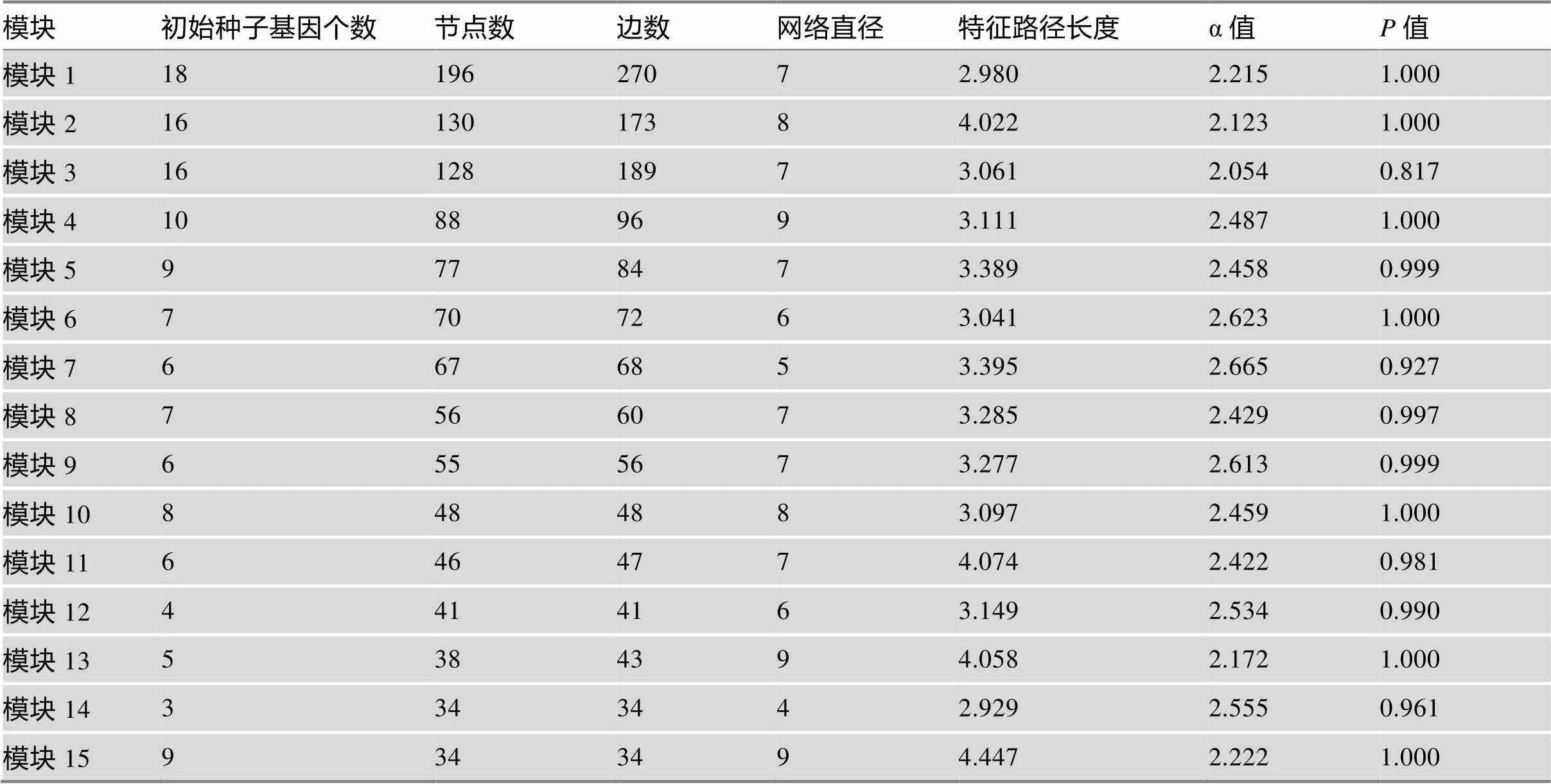
表1 各模块的基本特征和拓扑属性
α值为估计的幂律分布的指数参数;P值为KS检验的P值。
利用泊松检验,设定显著性水准为FDR<0.01,共得到58个核心基因。通过检索PubMed数据库,本研究发现共有34个核心基因被报道与NPC或OSCC相关,它们直接或间接参与了NPC和OSCC的发生发展,如增殖细胞核抗原(proliferating cell nuclear antigen, PCNA)、周期蛋白依赖激酶1 (cyclin- dependent kinase 1,CDK1)、信号转导与转录激活因子1 (signal transducer and activator of transcription 1, STAT1)、趋化因子CCL5 (C-C motif chemokine ligand 5, CCL5)等蛋白的编码基因。其余24个核心基因暂无文献报道与NPC或OSCC相关,这些基因可能对疾病的发生发展提供新的预测方向,如母体胚胎亮氨酸拉链激酶(maternal embryo leucine zipper kinase, MELK)、非转移性细胞蛋白1 (non-metastatic cell protein 1, NME1)、天冬氨酸特异性半胱氨酸蛋白酶-6 (caspase-6, CASP6)、RacGCT酶活性蛋白-1 (RACGAP1)、抑制素β A亚基基因(inhibin subunit beta A, INHBA)等蛋白的编码基因。核心基因数占模块中的初始种子基因数的44.6%。含有核心基因最多的模块是M2模块,共有10个核心基因。而含有核心基因最少的模块是M10模块,仅有含有1个核心基因(表2)。
2.5 模块的功能富集分析
KEGG通路富集分析结果显示,除了模块M6、M9、M10、M13、M14、M15外,其余9个模块至少富集到一条生物学通路(表3)。这些模块的显著富集于ECM受体相互作用(hsa04512)、黏着斑(hsa04510)、细胞周期(hsa04110)、P53信号通路(hsa04115)、PI3K-Akt信号通路(hsa04151)、细胞因子—细胞因子受体相互作用(hsa04060)等45个有统计学意义的相关通路(FDR值<0.001)。本研究根据富集到的KEGG通路类别评估了这9个功能模块之间的相互作用。为了清楚地展示各模块之间的关系,本研究用Cytoscape软件画出模块和通路分类关系图(见图3)。图中的功能类是KEGG通路的分类:信号转导(signal transduction);信号分子和相互作用(signaling molecules and interaction);细胞运动(cell motility);细胞生长和死亡(cell growth and death);复制与修复(replication and repair);细胞通讯(cell communication);免疫系统(immune system);内分泌系统(endocrine system);癌症(cancer);心血管疾病(cardiovascular diseases);发育(development);消化系统(digestive system);折叠、分类和降解(folding, sorting and degradation);传染病(infectious diseases)。其中细胞生长和死亡(M1、M2、M5和M8)和信号转导(M3、M4、M7和M11)连通度最高,体现了这些功能与NPC和OSCC的发病机制有密切的联系。本研究的结果表明,各模块之间不是孤立的,功能相一致的模块之间相互作用,共同影响疾病的发生发展。
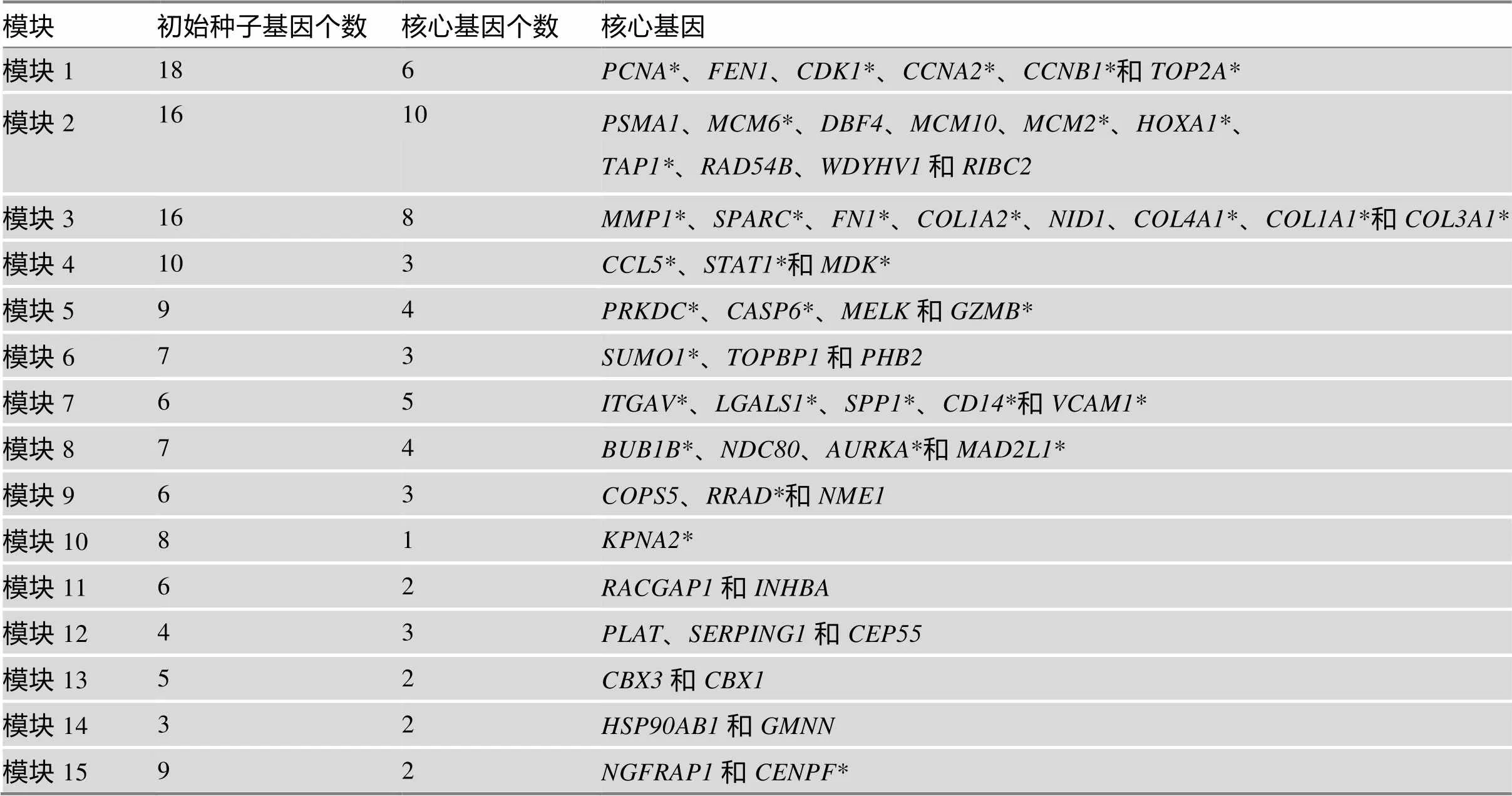
表2 各个模块的核心基因
*已有文献报道该基因与鼻咽癌和口腔鳞癌相关。
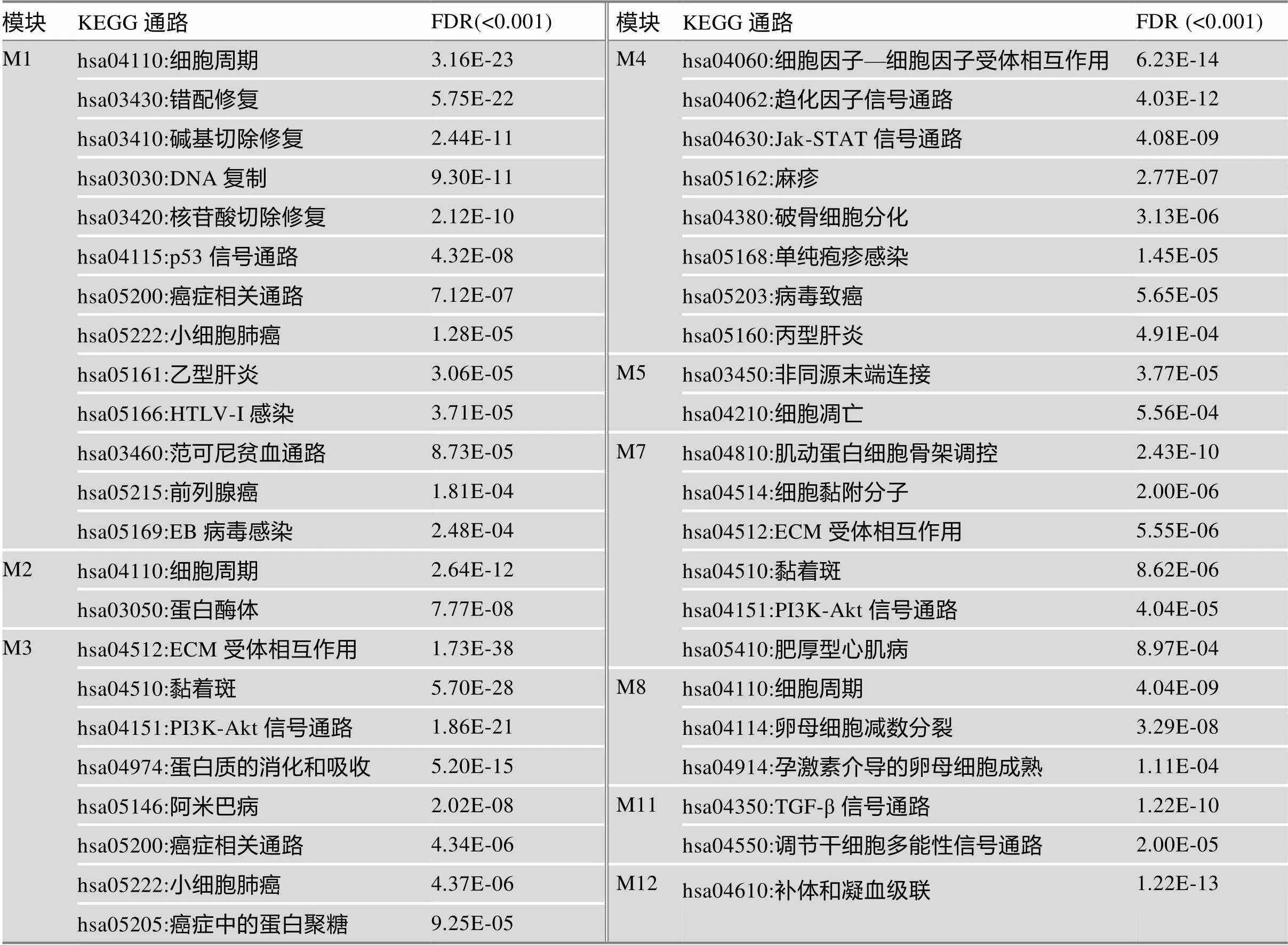
表3 各个模块的KEGG通路分析结果
3 讨论
基因多效性是癌症遗传机制中的普遍现象,大量全基因组关联研究(genome-wide association study, GWAS)的结果已经证实不同癌症表型可共享风险基因,即单个基因的突变可能引起不同的病理效应而参与多种癌症的发生发展[9]。基于基因多效性,本研究从GEO数据库分别下载NPC和OSCC基因芯片数据进行差异基因分析,以PPI知识为向导,两种癌症的共同差异表达基因为种子基因,构建NPC与OSCC共享风险基因网络。本研究利用Newman算法对NPC与OSCC共享风险基因网络进行网络分解,挖掘网络中高度模块化的共享功能模块和核心基因。通过阐明功能模块行使的生物学功能来挖掘NPC与OSCC的共享通路。
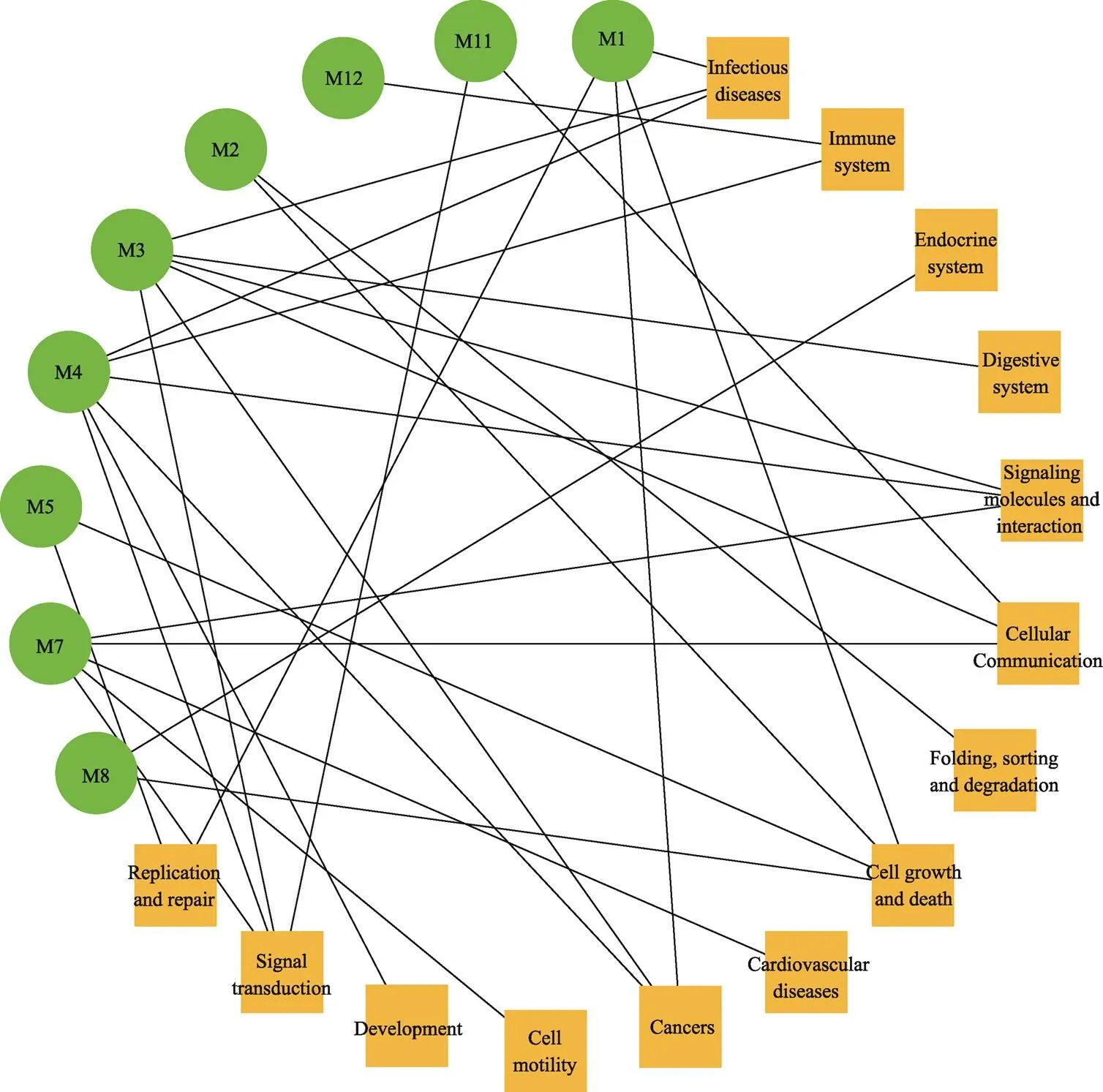
图3 共享功能模块与通路的关系图
在本研究中,共提取了15个NPC与OSCC相关的功能模块,并通过包括直径、特征路径长度、连通度等拓扑属性指标评价各模块的拓扑性质,结果显示各个模块都满足无标度性。模块中的核心基因担负着维系模块中各基因间相互联系和模块结构稳定性的作用,在模块行使特定生物学功能的过程中肩负着功能核心的作用,是功能模块必不可少的关键基因。通常,研究模块中核心基因的功能有助于阐明模块的生物学功能。
多数核心基因,如、、STAT1和等基因均被报道直接或间接与NPC和OSCC相关。它们可能直接或间接参与了肿瘤的生长和分化,如癌细胞的增殖、血管形成,侵袭和转移等。例如,基因的表达在真核细胞DNA复制的调控中起着至关重要的作用,是与多种肿瘤发生发展密切相关的基因。有研究表明,靶向基因的小干扰RNA (siRNA)通过沉默基因,可有效抑制对NPC细胞增殖的作用[10]。最近研究发现,高度侵袭性肿瘤表现出突变和活性,从而提高OSCC侵袭前的增殖能力[11],可用于评估异型增生和不同级别口腔鳞癌的增殖能力和侵袭性[12]。还有研究称,PCNA可以成为区分正常口腔粘膜和口腔鳞癌组织的生物标志物[13]。通过调节细胞中心体周期和有丝分裂的发生,在真核细胞周期的调控中发挥关键作用。目前有文献报道,通过草酸盐对乳酸脱氢酶(lactate dehydrogenase, LDH)的抑制,调控CDK1/cyclinb1通路,从而诱导G2/M细胞周期阻滞。CDK1/cyclin B1通路有望成为NPC治疗的靶点[14]。Chen等[15]报道发现,在OSCC中高表达,可能与OSCC的恶性程度有关,可作为判断OSCC恶性程度和预测预后的有用指标。可能在抑制肿瘤细胞生长,促进免疫监视、凋亡和细胞周期阻滞方面发挥关键作用[16]。最近有研究报道,表达下调导致细胞周期S期减少,G2/M期增加,有助于提高NPC细胞的放射敏感性[17]。有研究表明,的活化主要见于分化程度较高的肿瘤,如果OSCC患者的肿瘤中保留了的表达,并表现出的活化,则对辅助化疗有较好的疗效[18]。近年来报道证实,能直接参与恶性肿瘤的发生发展,在前列腺癌[19]、结肠癌[20]、肺癌[21]、乳腺癌[22]中都有过表达,并被证明通过不同的信号途径促进多种癌症类型的肿瘤细胞的生长和(或)运动。CCL5通过PI3K/AKT和HIF-1a途径诱导肿瘤血管生成,与人鼻咽癌血管生成密切相关,是NPC潜在的治疗靶点[23]。在慢性炎症状态下CCL5能激活VEGF和(或) STAT信号通路,促进细胞增殖和血管生成,可通过激活STAT和NF信号通路促进肿瘤形成[24]。有研究证实,CCL5通过其特异性高亲和力受体CCR5的诱导,增加OSCC的移动力[25]。
此外,本研究还发现了一些鲜有报道的与NPC和OSCC相关的核心基因,例如、、和等基因。这些核心基因在其他肿瘤的发生发展中起到重要作用,可能与NPC和OSCC也存在密切相关。例如,高表达,与肝癌[26]、肾上腺皮质癌[27]、乳腺癌[28]、胃癌[29]和脑癌[30]患者预后不良相关。在肝癌中高表达,可通过FOXM1/b-catenin信号通路调控细胞周期相关基因和有丝分裂进程,沉默可以降低肝癌细胞的活力和增殖,诱导细胞凋亡和有丝分裂,的稳定沉默能抑制肝癌细胞的侵袭、干性和致瘤性[26]。有报道指出,基因的编码蛋白被认为是潜在的典型转录因子,通过其DNA结合活性来调节基因转录与表达,与肿瘤细胞分化和转移抑制功能等高度相关。此外,NME1也参与DNA修复,这表明NME蛋白的表达减少可能导致基因组不稳定,从而导致癌症的发展[31]。具有调节细胞质分裂和细胞分化的作用。有文献报道,能激活RhoA和ERK蛋白,增强卵巢上皮癌细胞的迁移和侵袭能力[32]。的过表达与直肠癌的进展和预后密切相关,其生物学和临床意义为判断淋巴结转移和预后不良提供了充分的证据[33]。编码TGF-β超家族蛋白,在不同类型的肿瘤中发挥着重要作用。在食管腺癌中高表达,通过启动子去甲基化和组蛋白乙酰化可促进细胞增殖[34],也与淋巴结转移有关[35]。
为了阐明每个模块的生物学功能,本研究利用DAVID在线软件作了基于KEGG信号通路的富集分析。这些模块显著富集于ECM受体相互作用(has04512)、黏着斑(hsa04510)、细胞周期(hsa04110)、p53信号通路(hsa04115)、PI3K-Akt信号通路(hsa04151)、细胞因子—细胞因子受体相互作用(hsa04060)等。其中最显著相关的信号通路是ECM受体相互作用(hsa04512, M3)。ECM在细胞形态、增殖、迁移、分化、凋亡和癌变等多个生物学过程中发挥着重要的作用[36]。黏着斑是介导细胞对ECM粘附的调节效应的亚细胞结构[37]。根据KEEG信号通路富集分析显示,本研究富集到的ECM受体相互作用和粘着斑通路有许多基因重叠,例如编码胶原蛋白、整合素和层粘连蛋白等基因,有密切的相互作用。这些相互作用有利于癌细胞的增殖、运动、分化和ECM代谢,同时抑制细胞死亡、平稳的极化生长和ECM的稳定性[38]。ECM受体相互作用和粘着斑信号通路对癌细胞有明显调控的作用。在p53信号通路中,抑癌基因通过诱导细胞周期阻滞、凋亡或衰老,在细胞对各种应激信号的反应中起着至关重要的作用[39]。的失活能使调控的靶基因异常增殖和(或)表达缺失,可使细胞周期G1/S检查点失控,细胞凋亡失控,导致细胞癌变[40]。另外,模块与模块之间不是孤立的,功能相一致的模块之间相互作用,共同影响疾病的发生发展。模块M1、M2、M5和M8与细胞生长与死亡有关,共同调控细胞周期。模块M3和M7主要富集于ECM受体相互作用与黏着斑信号通路,共同协调参与癌细胞的增殖与迁移。M4模块主要富集于细胞因子—细胞因子受体相互作用、趋化因子信号通路和Jak-STAT信号通路,模块M11主要富集于TGF-β信号通路,M12主要富集于补体与凝血级联信号通路,这些信号通路都参与癌症的炎症反应,说明模块M4、M11和M12之间有密切联系。
总而言之,本研究通过对NPC和OSCC芯片数据的挖掘以及生物信息学分析,筛选出15个共享功能模块,58个密切相关的核心基因(如、、和等)以及p53信号通路、ECM受体相互作用、黏着斑等在NPC和OSCC的发生发展、侵袭、转移可能起着重要作用的信号通路,为研究NPC与OSCC的共享发病机制、肿瘤标志物及治疗靶点的筛选方面提供了更多的信息,有助于解释多效性基因在致癌过程中所起的作用。除此之外,本研究还发现了一些鲜有报道的核心基因,如、、和等,这些新发现为探索NPC与OSCC的共同发生发展机制提供了新线索。综上所述,本研究表明鼻咽癌和口腔鳞癌具有相似的致癌机制,所挖掘的共享模块可能是这两种疾病演化的核心分子相互作用机制。
[1] Chua MLK, Wee JTS, Hui EP, Chan ATC. Nasopharyngeal carcinoma., 2016, 387(10022): 1012–1024.
[2] Jiang S, Dong Y. Human papillomavirus and oral squamous cell carcinoma: A review of HPV-positive oral squamous cell carcinoma and possible strategies for future., 2017, 41(5): 323–327.
[3] Stearns FW. One hundred years of pleiotropy: a retrospective., 2010, 186(3): 767–773.
[4] Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects., 2018, 19(9): 547–562.
[5] Newman ME. Modularity and community structure in networks., 2006, 103(23): 8577–8582.
[6] Clauset A, Newman ME, Moore C. Finding community structure in very large networks., 2004, 70(6 pt 2): 66111.
[7] Zhao XL, Zuo XY, Qin JH, Liang Y, Zhang NZ, Luan YZ, Rao SQ. A novel biological pathway expansion method based on the knowledge of protein-protein interactions.,2014, 36(04): 387–394.赵小蕾, 左晓宇, 覃继恒, 梁岩, 张乃尊, 栾奕昭, 饶绍奇. 基于蛋白质互作知识的生物学通路扩充新方法. 遗传, 2014, 36(4): 387–394.
[8] Travers J, Milgram S. An experimental study of the small world problem., 1969, 32(4): 425–443.
[9] Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies., 2013, 14(7): 483–495.
[10] Wang J, Mei F, Gao X, Wang S. Identification of genes involved in Epstein-Barr virus-associated nasopharyngeal carcinoma., 2016, 12(4): 2375–2380.
[11] Kato K, Kawashiri S, Yoshizawa K, Kitahara H, Okamune A, Sugiura S, Noguchi N, Yamamoto E. Expression form of p53 and PCNA at the invasive front in oral squamous cell carcinoma: correlation with clinicopathological features and prognosis., 2011, 40(9): 693–698.
[12] Poosarla C, Ramesh M, Ramesh K, Gudiseva S, Bala S, Sundar M. Proliferating cell nuclear antigen in premalignancy and oral squamous cell carcinoma., 2015, 9(6): ZC39–C41.
[13] Madan M, Chandra S, Raj V, Madan R. Evaluation of cell proliferation in malignant and potentially malignant oral lesions., 2015, 19(3): 297–305.
[14] Zhai X, Yang Y, Wan J, Zhu R, Wu Y. Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells., 2013, 30(6): 2983–2991.
[15] Chen X, Zhang FH, Chen QE, Wang YY, Wang YL, He JC, Zhou J. The clinical significance of CDK1 expression in oral squamous cell carcinoma., 2015, 20(1): e7–e12.
[16] Zhang Y, Liu Z. STAT1 in cancer: friend or foe?, 2017, 24(130): 19–29.
[17] Qu S, Guo Y, Huang ST, Zhu XD. Inhibition of STAT1 sensitizes radioresistant nasopharyngeal carcinoma cell line CNE-2R to radiotherapy., 2018, 9(9): 8303–8310.
[18] Laimer K, Spizzo G, Obrist P, Gastl G, Brunhuber T, Schafer G, Norer B, Rasse M, Haffner MC, Doppler W. STAT1 activation in squamous cell cancer of the oral cavity: a potential predictive marker of response to adjuvant chemotherapy., 2007, 110(2): 326–333.
[19] Urata S, Izumi K, Hiratsuka K, Maolake A, Natsagdorj A, Shigehara K, Iwamoto H, Kadomoto S, Makino T, Naito R, Kadono Y, Lin WJ, Wufuer G, Narimoto K, Mizokami A. C-C motif ligand 5 promotes migration of prostate cancer cells in the prostate cancer bone metastasis microenvironment., 2018, 109(3): 724–731.
[20] Kan JY, Wu DC, Yu FJ, Wu CY, Ho YW, Chiu YJ, Jian SF, Hung JY, Wang JY, Kuo PL. Chemokine (C-C motif) ligand 5 is involved in tumor-associated dendritic cell- mediated colon cancer progression through non-coding RNA MALAT-1., 2015, 230(8): 1883–1894.
[21] Huang CY, Fong YC, Lee CY, Chen MY, Tsai HC, Hsu HC, Tang CH. CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways., 2009, 77(5): 794–803.
[22] Khalid A, Wolfram J, Ferrari I, Mu C, Mai J, Yang Z, Zhao Y, Ferrari M, Ma X, Shen H. Recent advances in discovering the role of CCL5 in metastatic breast cancer., 2015, 15(13): 1063–1072.
[23] Ma W, Feng L, Zhang S, Zhang H, Zhang X, Qi X, Zhang Y, Feng Q, Xiang T, Zeng YX. Induction of chemokine (C-C motif) ligand 5 by Epstein-Barr virus infection enhances tumor angiogenesis in nasopharyngeal carcinoma., 2018, 109(5): 1710–1722.
[24] Rao SK, Pavicevic Z, Du Z, Kim JG, Fan M, Jiao Y, Rosebush M, Samant S, Gu W, Pfeffer LM, Nosrat C A. Pro-inflammatory genes as biomarkers and therapeutic targets in oral squamous cell carcinoma., 2010, 285(42): 32512–32521.
[25] Chuang JY, Yang WH, Chen HT, Huang CY, Tan TW, Lin YT, Hsu CJ, Fong YC, Tang CH. CCL5/CCR5 axis promotes the motility of human oral cancer cells., 2009, 220(2): 418–426.
[26] Xia H, Kong SN, Chen J, Shi M, Sekar K, Seshachalam VP, Rajasekaran M, Goh B, Ooi LL, Hui KM. MELK is an oncogenic kinase essential for early hepatocellular carcinoma recurrence., 2016, 383(1): 85–93.
[27] Kiseljak-Vassiliades K, Zhang Y, Kar A, Razzaghi R, Xu M, Gowan K, Raeburn CD, Albuja-Cruz M, Jones KL, Somerset H, Fishbein L, Leong S, Wierman ME. Elucidating the role of the maternal embryonic leucine zipper kinase in adrenocortical carcinoma., 2018, 159(7): 2532–2544.
[28] Speers C, Zhao SG, Kothari V, Santola A, Liu M, Wilder-Romans K, Evans J, Batra N, Bartelink H, Hayes DF, Lawrence TS, Brown PH, Pierce LJ, Feng FY. Maternal embryonic leucine zipper kinase (MELK) as a novel mediator and biomarker of radioresistance in human breast cancer., 2016, 22(23): 5864–5875.
[29] Li S, Li Z, Guo T, Xing XF, Cheng X, Du H, Wen XZ, Ji JF. Maternal embryonic leucine zipper kinase serves as a poor prognosis marker and therapeutic target in gastric cancer., 2016, 7(5): 6266–6280.
[30] Liu H, Sun Q, Sun Y, Zhang J, Yuan H, Pang S, Qi X, Wang H, Zhang M, Zhang H, Yu C, Gu C. MELK and EZH2 cooperate to regulate medulloblastoma cancer stem- like cell proliferation and differentiation., 2017, 15(9): 1275–1286.
[31] Puts GS, Leonard MK, Pamidimukkala NV, Snyder DE, Kaetzel DM. Nuclear functions of NME proteins., 2018, 98(2): 211–218.
[32] Wang C, Wang W, Liu Y, Yong M, Yang Y, Zhou H. Rac GTPase activating protein 1 promotes oncogenic progression of epithelial ovarian cancer., 2018, 109(1): 84–93.
[33] Imaoka H, Toiyama Y, Saigusa S, Kawamura M, Kawamoto A, Okugawa Y, Hiro J, Tanaka K, Inoue Y, Mohri Y, Kusunoki M. RacGAP1 expression, increasing tumor malignant potential, as a predictive biomarker for lymph node metastasis and poor prognosis in colorectal cancer., 2015, 36(3): 346–354.
[34] Seder C W, Hartojo W, Lin L, Silvers A L, Wang Z, Thomas D G, Giordano T J, Chen G, Chang AC, Orringer MB, Beer DG. INHBA overexpression promotes cell proliferation and may be epigenetically regulated in esophageal adenocarcinoma., 2009, 4(4): 455–462.
[35] Lyu S, Jiang C, Xu R, Huang Y, Yan S. INHBA upregulation correlates with poorer prognosis in patients with esophageal squamous cell carcinoma., 2018, 10: 1585–1596.
[36] Hansen NU, Genovese F, Leeming DJ, Karsdal MA. The importance of extracellular matrix for cell function andlikeness., 2015, 98(2): 286–294.
[37] Wu C. Focal adhesion: a focal point in current cell biology and molecular medicine., 2007, 1(1): 13–18.
[38] He Y, Shao FY, Pi WD, Shi C, Chen YJ, Gong DP, Wang BJ, Cao ZW, Tang KL. Largescale transcriptomics analysis suggests over-expression of BGH3, MMP9 and PDIA3 in oral squamous cell carcinoma., 2016, 11(1): e146530.
[39] Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway., 2009, 9(12): 862–873.
[40] Gong Z, Yang Q, Zeng Z, Zhang W, Li X, Zu X, Deng H, Chen P, Liao Q, Xiang B, Zhou M, Li X, Li Y, Xiong W, Li G. An integrative transcriptomic analysis reveals p53 regulated miRNA, mRNA, and lncRNA networks in nasopharyngeal carcinoma., 2016, 37(3): 3683–3695.
Shared functional modules for nasopharyngeal and oral squamous cell carcinoma identified by network analysis of transcriptomes
Yingjian Chen1,2, Yuanjun Liao1,2, Fan Lin1,2, Shengnan Sun1,2, Xiaolei Zhao2, Jiheng Qin2, Shaoqi Rao2
Although nasopharyngeal carcinoma (NPC) and oral squamous cell carcinoma (OSCC) are highly correlated clinical diseases, the underling molecular mechanisms to link the two diseases remain largely unknown. The aim of this study is to identify the shared functional modules for NPC and OSCC by using large-scale transcriptomic data. Gene expression profile datasets of NPC and OSCC were obtained from the GEO database. A total of 1279 differentially expressed genes (DEGs) of NPC and 1293 DEGs of OSCC were identified by fold change and empirical Bayes method, and 278 DEGs were common to these two diseases. These overlapped genes were translated into a primary network consisting of 1290 nodes (genes) and 1766 edges. The primary network was then decomposed into 15 compacted modules (subnets) with high modularity by Newman’s algorithm. Topological analysis of these modules identified a total of 58 hub genes, most of which (e.g.,,,,and) have been proved to be associated with NPC and/or OSCC, while the rest (e.g.,,,,, and) might be novel risk genes for the two diseases. Further bioinformatics analysis of KEGG databases revealed that these modules are involved in multiple pathogenic biological pathways for either NPC or OSCC (e.g., p53 signaling pathway, ECM-receptor interaction, focal adhesion, and cell cycle). This study demonstrates that NPC and OSCC have similar molecular bases, and the identified pleiotropic modules may shape the complicated molecular interplays underlying the two clinically correlated diseases.
nasopharyngeal carcinoma; squamous cell carcinoma; microarrays; network analysis; pleiotropism
2018-09-07;
2018-10-26
国家自然科学基金项目(编号:81373085) 资助[Supported by the National Natural Science Foundation of China (No. 81373085)]
陈应坚,硕士研究生,专业方向:公共卫生。E-mail: 1053879830@qq.com
饶绍奇,教授,博士生导师,研究方向:遗传统计与生物信息学。E-mail: raoshaoq@gdmu.edu.cn
10.16288/j.yczz.18-215
2019/1/16 9:14:53
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190116.0914.002.html
(责任编委: 周钢桥)
