染色体显性遗传性多囊肾病PKD2基因突变的检测
2019-01-16陈艳黄翀徐承云
陈艳 黄翀 徐承云


【摘要】 目的:建立检测常染色体显性遗传性多囊肾病2型(polycystic kidney disease 2,PKD2)致病基因突变的方法,检测收集的10个ADPKD家系共15例患者的PKD2基因突变情况。方法:收集江西地区经临床确诊的常染色体显性遗传性多囊肾病患者15例,采集外周静脉血5 mL,用试剂盒提取基因组DNA,采用聚合酶链式反应(PCR)扩增PKD2基因全部外显子及相近内含子区域,PCR扩增产物经分离纯化后直接进行基因序列测序,根据测序图谱进行突变分析,明确基因突变位点和类型。结果:15例常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)患者中,检测出3个正常基因多态性位点,分别为1号外显子第420位碱基G置换为A,未引起编码氨基酸改变;1号外显子第568位碱基G置换为A,致使190位编码氨基酸由丙氨酸改变为苏氨酸;内含子靠近cDNA第844位碱基5端的第22个碱基A置换为G。结论:可通过直接基因序列测定对PKD2进行基因突变检测,本研究所检测到的3个正常基因多态性位点均已有报道,为开展ADPKD直接基因诊断、产前诊断及症状前诊断提供了实验基础。
【关键词】 常染色体显性遗传性多囊肾病; PKD2; 基因突变; 多态性
Detection of PKD2 Gene Mutation in Chromosomal Dominant Polycystic Kidney Disease/CHEN Yan,HUANG Chong,XU Chengyun.//Medical Innovation of China,2019,16(26):-150
【Abstract】 Objective:To set up a method for detecting the mutations of autosomal dominant olycystic kidney disease gene 2, detect the mutations of PKD2 in 15 patients from 10 ADPKD families.Method:Fifteen patients with autosomal dominant polycystic kidney disease diagnosed clinically in Jiangxi were collected,5 mL peripheral venous blood was collected and genomic DNA was extracted with the kit.All exons and close intron regions of PKD2 gene were amplified by polymerase chain reaction(PCR),after isolation and purification,PCR amplification products were directly sequenced,and mutation analysis was conducted according to the sequencing map to identify the mutation sites and types of genes.Result:Three polymorphisms were detected in 15 ADPKD patients.The c.568G>A in exon1 caused the translation change(Ala190Thr).The other two (c.420G>A in exon1 and 844-22A>G in IVS3) did not cause the translation change.Conclusion:Gene mutation detection of PKD2 can be performed by direct gene sequencing,all the 3 normal gene polymorphisms detected in this study have been reported, providing an experimental basis for direct gene diagnosis, prenatal diagnosis and pre-symptom diagnosis of ADPKD.
【Key words】 Autosomal dominant polycystic kidney disease; PKD2; Gene mutation; Polymorphism
First-authors address:Second Affiliated Hospital of Nanchang University,Nanchang 330006,China
doi:10.3969/j.issn.1674-4985.2019.26.039
常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)又称为成人型多囊肾病,是最常见的肾脏遗传病,发病率为1/400~1/1 000[1],我国约有150万例患者。ADPKD的主要病理特征为双侧肾脏出现无数大小不等的液性囊泡,并随着年龄的增长囊肿进行性长大。约50%的ADPKD患者在60岁时发展至终末期肾病(end stage renal disease,ESRD)[1]。ADPKD所致的透析患者约占整个透析人群的4.4%[2]。ADPKD可累及人体多个组织器官,除肾脏改变外,还可出现肝脏囊肿、胰腺囊肿、脾脏囊肿、高血压、颅内动脉瘤、心瓣膜病、左心室肥大及结肠憩室等肾外病变。ADPKD是一种单基因遗传性疾病,目前研究发现ADPKD的致病基因有3个,分别为PKD1、PKD2和PKD3。PKD1基因定位于人染色体16p13.3,PKD2是一个单拷贝基因,定位于人染色体4q22-23,有15个外显子;PKD3基因仅数个家系中出现,目前报道较少,尚未定位克隆。PKD1基因突变引起的ADPKD患者占85%,约15%的ADPKD患者为PKD2突变所致[3]。ADPKD具有遗传显性延迟性,患者在發病前可能已经将致病的基因突变遗传给了下一代,且目前尚无特效治疗方法。因此对ADPKD家系中存在发病风险的成员早期进行症状前基因诊断,对有高发病风险胎儿或胚胎进行产前诊断及植入前基因诊断显得很有意义。目前已报道的PKD2基因突变散布于PKD2基因的各个区域,无突变热点,且大多数基因突变为单发;以上报道大多集中在高加索人种。为进一步了解我国PKD2基因突变的发生情况,研究其突变规律,本研究通过直接序列测定(direct sequencing,DS),对江西地区收集的10个ADPKD家系共15例患者进行PKD2全基因编码区突变检测,为开展ADPKD直接基因诊断、产前诊断及症状前诊断提供了实验基础。
1 资料与方法
1.1 一般资料 收集江西地区10个ADPKD家系共15例经临床确诊的ADPKD患者,纳入研究,编号p1-p15,完成多囊肾病例调查表,询问家族史并绘制家系图。诊断标准:有阳性家族史,15~39岁,双侧肾脏囊肿数≥3个;40~59岁,每侧肾脏囊肿数≥2个;年龄≥60岁,每侧肾脏囊肿数≥4个;排除标准:40岁以上,如果双侧肾脏囊肿数<2个,即可排除[1]。患者均知情同意,且该研究经医院伦理委员会批准。
1.2 方法 所有纳入研究的对象均收集外周静脉血5 mL,EDTA抗凝,分选白细胞,提取基因组DNA[QuickGene DNA whole blood kit S(DB-S)];通过聚合酶链式反应(PCR)特异扩增PKD2基因全部外显子及相近内含子区域,PCR扩增产物经分离纯化后直接进行基因序列测序;运用Chromas软件查看测序结果及DNAMAN软件进行多重序列结果比对。
通过美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)获得PKD2基因组序列,Genebank序号为NG_008604.1,使用Primer primier5.0软件设计PKD2基因15个外显子特异的PCR扩增引物,其中Exon 1长585 bp,由于其GC含量高,需进行分段扩增,故相互交错设计3对引物,覆盖整个外显子。通过进行PCR预实验摸索出最佳的PCR反应条件,见表1。
2 结果
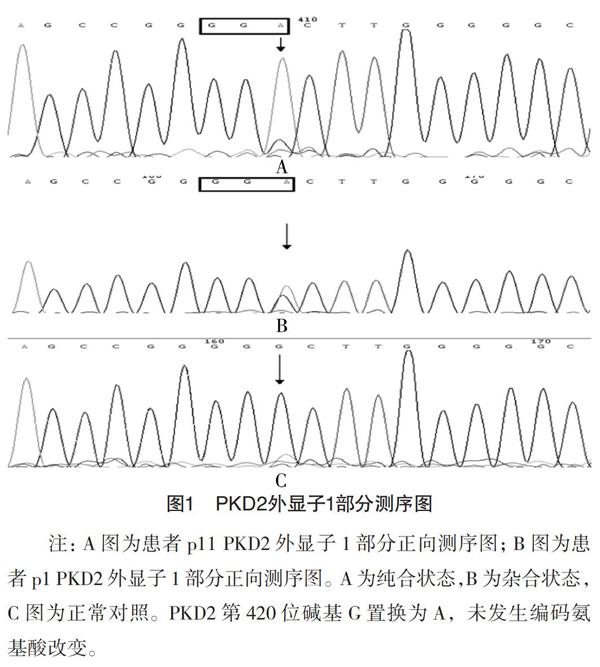
在15例ADPKD患者中,检测出3个PKD2正常基因多态性位点,见表2。7例患者PKD2基因的1外显子测序显示cDNA第420位碱基G置换为A(图1),未编码氨基酸改变;其中4例以杂合状态发生,3例以纯合状态发生。10例患者PKD2基因的1外显子测序显示cDNA第568位碱基G置换为A(图2),致使190位编码氨基酸由丙氨酸改变为苏氨酸,其中6例以杂合状态发生,4例以纯合状态发生。2例患者内含子区域靠近cDNA第844位碱基5端的第22个碱基A置换为G(图3),不影响编码区的特异性剪切;均以杂合状态发生。以上3个正常基因多态性位点均已被报道。
3 讨论
ADPKD是一种单基因遗传性疾病,具有遗传异质性(genetic heterogeneity)。目前研究发现ADPKD的致病基因有3个,分别为PKD1、PKD2和PKD3。PKD1基因突变引起的ADPKD患者占85%,约15%的ADPKD患者为PKD2突变所致。PKD1及PKD2均已成功定位和克隆[4];PKD3基因仅数个家系中出现,目前报道较少,尚未定位克隆。目前PKD1及PKD2均已成功定位和克隆[4];PKD3基因目前报道较少,尚未定位克隆。
PKD1基因定位于人染色体16p13.3,有46个外显子,基因序列长约52 kb,转录的mRNA长14.1 kb,编码含有4 302个氨基酸的多囊蛋白-1(polycystin-1,PC-1)。PKD1基因5端的第1-33外显子含重复序列,属于多拷贝区[5];第34~46外显子为单拷贝区。PKD1基因序列中GC含量较高,且人基因组内存在6个与PKD1基因有97%~99%相似度的同源序列区,这些都显著增加了基因检测的难度。PC-1是一种分布于细胞膜的糖蛋白,有11个跨膜区域。PC1的氨基端位于细胞外,而羧基端位于细胞内,约75%的蛋白结构位于细胞外。PC1各区域的功能尚未完全研究清楚。
PKD2是一个单拷贝基因,定位于人染色体4q22-23,有15个外显子,基因长68 kb,转录RNA长5.4 kb,编码含968个氨基酸的多囊蛋白-2(polycystin-2,PC-2)。PC-2是一种非选择性的钙离子通道[6]。PC-2含有6个与PC-1跨膜区域结构部分相似的跨膜区,其氨基端及羧基端均位于细胞内。文献[7]研究表明,PC-2的第1-223位氨基酸为氨基端的胞内区,490-505位、572-598位及680-968位为胞内区,224-244位、469-489位、506-526位、551-571位、599-619位及659-679位为跨膜区,245-468位、527-550位及620-658位为胞外区。其中763-796位为螺旋区-螺旋区,此区被认为是PC-2与PC-1结合的区域,763-774位为钙离子结合区域(EFhand)。
PC-2与PKD1编码产物多囊蛋白-1(PC-1)之间相互作用,在肾小管上皮细胞的初级纤毛上形成多囊蛋白复合物,调节上皮细胞的增殖和分化。纤毛致病学说认为此多囊蛋白复合物是一种机械性刺激感受器,能感受肾小管腔内的液压变化,通过TRPP2通道控制Ca2+的内流,从而影响细胞内Ca2+和cAMP的水平。腺苷酸环化酶和酪氨酸激酶受体途径间的异常干扰,加上细胞内Ca2+浓度改变,促进了肾小管上皮细胞的增殖和液体分泌,最终导致ADPKD的发生。
梅奥医学中心是美国乃至全球研究PKD最领先的科研机构,Paavola等联合多家中心组建了PKD突变基因网络数据库PKDB(http://pkdb.pkdcure.org),至今已录入2 080个独立家系ADPKD突变基因和2 323个突变位点[8-9]。PKDB可通过邮件接收世界范围内PKD突变基因测序结果,再通过审查并录入PKD1和PKD2多态性变异以及其他PKD相关的基因变异。与国际发展趋势相比,我国建立PKD网络数据库受到临床诊治水平参差不齐的制约,PKD登记和注册网络数据库目前尚未建立,各中心之间无协作网络。由于在全国范围内没有广泛开展基因诊断,缺乏确诊手段和技术,导致PKD确诊率低,不少家系误诊漏诊和不规范治療。
ADPKD基因突变的种类很多,包括有同义突变(synonymous mutation)、无义突变(nonsense mutation)、错义突变(missense mutation)、插入和缺失突变(deletion and insertion),移码突变(frameshift mutation)、剪切突变(splice),内含子沉默突变(intervening sequence silent variations,IVS silent)等。不影响编码区特异性剪切的内含子突变及同义突变为基因多态性。
目前国内外已报道的PKD2基因突变报道大多集中在高加索人种,其中包含无义突变、移码突变、剪切突变、插入或缺失突变、错义突变、同义突变及内含子突变[10]。这些基因突变散布于PKD2基因的各个区域,无突变热点,且大多数基因突变为单发。无义突变或移码突变可使碱基发生改变,产生终止密码子,可造成基因编码产物多囊蛋白的截短或缺失,使其失去正常的功能;按照Germino的标准[11],可确定其为与致病相关的基因突变。由于缺乏对多囊蛋白的确切的功能认识,PCR扩增产物中检测出的错义或剪切突变,若基因突变位于推测的多囊蛋白重要功能区域或突变可能对编码区的剪接造成影响,且突变仅与患者伴随出现,则可高度怀疑此突变与致病相关。
ADPKD患者与普通人群一样存在基因多态性。尽管本研究未选取健康人群为对照,但本研究检测到的3个基因多态性位点c.420G>A、c.568G>A、844-22A>G均已被报道证实为正常基因多态性位点[12-14]。
虽然ADPKD具体的分子生物学致病机制尚不明确[15-17],但“二次打击”学说(即体细胞等位基因突变学说)被广大学者普遍接受。“二次打击”学说认为:囊肿的形成不仅需要从生殖细胞遗传获得PKD1或PKD2(两者为等位基因)其中一个的突变基因,且需要体细胞在发育过程中在后天局部因素的作用下受到二次打击(即丢失了正常的单倍体)。第一次突变是从患病父母遗传来的胚系突变,存在于所有的细胞中,包括肾小管上皮细胞。第一次突变是必要的,但不足以导致囊肿的形成。第二次打击,是指在个体的肾小管体细胞中发生突变,使正常的PKD1或PKD2的等位基因失活或突变,才能最终导致肾小管上皮细胞的异常增殖和囊肿形成[18-20]。第二次体细胞突变可以是同一致病基因的纯合性突变,也可以是PKD1或PKD2不同致病基因之间的杂合性突变。每一个肾囊肿都是体细胞基因的第二次突变引发的克隆性增殖。ADPKD患者肾囊肿只发生于部分肾单位,约占全部肾单位的5%。目前尚无方法可预知第二次打击。
随着分子诊断技术的不断进步,基因诊断的检出率及准确率大大提高,检测费用也逐渐降低至可接受范围,使得ADPKD患者基因诊断的可行性不断升高。但由于不同地区及不同民族间ADPKD发病情况存在差异,应积极建立适合我国人种的简便、敏感及准确性高的PKD基因诊断体系。本研究在江西地区ADPKD患者中检测到3个正常基因多态性位点,为开展ADPKD直接基因诊断、产前诊断及症状前诊断提供了实验基础。此外还应进一步扩大受检家系,完善PKD1基因突变检测,优化检测体系,提高突变检出率,争取早日实现ADPKD症状前诊断和产前诊断,及早防治并发症,提高ADPKD患者生存质量。
参考文献
[1]常染色体显性多囊肾病临床实践指南专家委员会.中国常染色体显性多囊肾病临床实践指南(第二版)[J].临床肾脏病杂志,2019,19(4):227-235.
[2]薛澄,周晨辰,许晶,等.基于常染色体显性多囊肾病基因芯片数据的生物信息学分析[J].协和医学杂志,2017,8(2):171-177.
[3] Chapma N,Arlene B.Autosomal dominant polycystic kidney disease (ADPKD):executive summary from a Kidney Disease:Improving Global Outcomes(KDIGO) Controversies Conference[J].Kidney International:Official Journal of the International Society of Nephrology,2015,1(1):17-27.
[4] Irazaba L,Maria V.Imaging Classification of Autosomal Dominant Polycystic Kidney Disease:A Simple Model for Selecting Patients for Clinical Trials[J].Journal of the American Society of Nephrology,2015,1(1):160-172.
[5] Dell K M,Matheson M,Hartung E A,et al.Kidney disease progression in autosomal recessive polycystic kidney disease[J].
J Pediatr,2016,171:196-201.
[6] Schrier R W,Abebe K Z,Perrone R D,et al.Blood Pressure in Early Autosomal Dominant Polycystic Kidney Disease[J].N Engl J Med,2014,371(24):2255-2266.
[7] Torres V E,Abebe K Z,Chapman A B,et al.Angiotensin Blockade in Late Autosomal Dominant Polycystic Kidney Disease[J].New England Journal of Medicine,2014,371(24):2267-2276.
[8]薛澄,周晨辰,梅長林.多囊肾病网络数据库的研究进展[J/CD].中华肾病研究电子杂志,2017,6(1):31-33.
[9] Paavola J,Schliffke S,Rossetti S,et al.Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy[J].J Mol Cell Cardiol,2013,58:199-208.
[10]刘杰,刘福颂,王芳,等.常染色体显性多囊肾病家系基因特点的研究[J].中华老年心脑血管病杂志,2018,20(9):936-939.
[11] Tan Y,Blumenfeld J R,Donahue S,et al.Novel method for genomic analysis of PKD1 and PKD2 mutations in autosomal dominant polycystic kidney disease[J].Human Mutation,2010,30(2):264-273.
[12] Garcia-Gonzalez M A.Evaluating the clinical utility of a molecular genetic test for polycystic kidney disease[J].Molecular Genetics & Metabolism,2007,92(1):160-167
[13]李新伟,张群芝.遗传性多囊肾家系PKD1、PKD2基因突变探讨与研究[J].中国优生与遗传杂志,2017,25(10):10-11.
[14]骆杰伟,孟晓嵘,郑星宇,等.常染色体显性遗传性多囊肾家系PKD1、PKD2基因突变分析[J].肾脏病与透析肾移植杂志,2014,(4):321-325,336.
[15] Verghese P,Miyashita Y.Neonatal polycystic kidney disease[J].Clinics in Perinatology,2014,41(3):543-560.
[16] Sans-atxer L,Torra R,Fern?ndez-llama P.Hypertension in autosomal -dominant polycystic kidney disease(ADPKD)[J].Clinical Kidney Journal,2013,5(5):457-460.
[17] Torra B R,Ars C E.Molecular diagnosis of autosomal dominant polycystic kidney disease[J].Nefrología Publicación Official De La Sociedad Espaola Nefrologia,2011,31(1):35-43.
[18] Hoefele J,Mayer K,Scholz M,et al.Novel PKD1 and PKD2 mutations in autosomal dominant polycystic kidney disease(ADPKD)[J].Nephrol Dial Transplant,2011,26(7):2181-2188.
[19] Paul B M,Vanden Heuvel G B.Kidney:polycystic kidney disease[J].Wiley Interdisciplinary Reviews Developmental Biology,2014,3(6):465-487.
[20]陳冬平.中国汉族人群常染色体显性多囊肾病的临床特征、危险因素及干预研究[D].上海:第二军医大学,2016.
(收稿日期:2019-06-28) (本文编辑:张爽)
