食用植物油中3-MCPD酯GC-MS测定方法研究
2019-01-15王永瑞徐宇丽魏长庆
王永瑞 王 丹 徐宇丽 魏长庆
(石河子大学食品学院1,石河子 832000)(新疆植物药资源利用教育部重点实验室2,石河子 832000)(石河子出入境检验检疫局3,石河子 832000)
新疆占中国1/6左右国土面积,地大物博,特产丰富,瓜果类农产品更是享誉国内外。近年来,利用新疆特有瓜果籽仁为原料生产的特种油脂不断深入市场,并赢得广大人民群众的好评[1]。目前,新疆市场销售的特种油脂有:红花籽油、葡萄籽油、番茄籽油等多个种类。在植物油脂中,不同种类的脂肪酸、维生素以及不同的天然色素和微量元素对人体起着重要的作用,特种油脂因其脂肪酸组成的特殊性对各种类型体质的人群起到不同的保健效果,越来越受到广大消费者青睐[2]。然而近年来研究发现,在各类食用油以及油类食品中存在着一种潜在的致癌物质—氯丙醇酯,这使得食用油的安全问题更加引人关注。
3-MCPD酯是4类氯丙醇酯中的1类,最早发现于1980年,由Velíšek等[3]在用酸水解蔬菜蛋白过程中发现。随着检测设备以及检测方法的发展,陆续有研究者在各类食品中检测出3-MCPD酯的存在,如饼干、咖啡、薯条、鱼油、食用油、婴幼儿奶粉等中都有存在[4-9]。3-MCPD酯之所以备受关注,不仅仅是因为其作为一种非营养物质的存在,而是因为其进入人体后会被水解为游离的3-MCPD。这种物质已知具有肾脏毒性,致癌性以及致突变等危害,世界各国对3-MCPD都有相应的限量标准[10-11]。2017年9月,随着新的国家食品安全标准GB 2762—2017《食品中污染物痕量》的正式实施,关于调味品中3-MCPD限量指标也有着明确的规定。因此,本研究采用间接法将油样中的3-MCPD酯经过甲醇钠-甲醇碱水解为3-MCPD,再固相萃取柱净化,七氟丁酰咪唑(HFBI)衍生结合GC-MS方法进行检测。利用该方法测定了新疆几种市售的常见食用植物油和几种特种食用植物油中3-MCPD酯的含量,评估新疆市售食用植物油中3-MCPD酯的风险水平的同时也为食用植物油中3-MCPD酯变化规律的研究提供参考。
1 材料与方法
1.1 材料与仪器
亚麻籽油、红花籽油、核桃油、花生油、番茄籽油、葡萄籽油、核桃油、菜籽油: (均为一级油,生产地均为新疆);3-MCPD标准品(纯度99.6%);氘代同位素d5-3-MCPD标准品(纯度98.5%);3-MCPD 棕 榈 酸 二 酯标准品 ( 98%),d5-3-MCPD 棕榈酸二酯标准品(纯度97%);N-七氟丁酰基咪唑(98%);甲基叔丁基醚、无水硫酸钠、冰醋酸:分析纯;甲醇钠:化学纯;乙酸乙酯、甲醇、正己烷:色谱纯。
7890A-5975C气质联用仪;85-2 恒温磁力搅拌器;EL204 电子天平;Xcelvap 全自动氮吹浓缩仪;Milli-Q 去离子水发生器;CleanertMCPD 氯丙醇专用固相萃取柱。
1.2 标准溶液的配制
准确称取 3-MCPD、d5-3-MCPD、3-MCPD 棕榈酸二酯、d5-3-MCPD 棕榈酸二酯各 10.0 mg (精确至 0.1 mg),分别置于4支10 mL棕色容量瓶中,加入乙酸乙酯配制成质量浓度为1 000 mg/L的标准液,于-20 ℃冰箱内冷藏储存。分别将单标储备液用乙酸乙酯稀释至 100 mg/L,作为标准中间液。使用时用正己烷逐级稀释100 mg/L的标准中间液至所需浓度。
1.3 样品前处理
1.3.1 水解与净化
水解:准确称取0.10 g油样至12 mL具盖玻璃试管中,加入50 μL 10 mg/L的d5-3-MCPD 棕榈酸二酯内标溶液(相当于 100 ng 游离态的 d5-3-MCPD),加入0.5 mL甲基叔丁基醚-乙酸乙酯(8∶2)混合液后,45 ℃下超声混匀。加入0.5 mL 甲醇钠-甲醇(0.5 mol/L)室温下水解4 min后,迅速加入3.0 mL冰乙酸溶液(1.0 mL冰乙酸溶于30 mL 20% NaCl溶液中),充分涡旋混匀。加入3.0 mL正己烷脱上层油相,涡旋静置。待明显分层后,去除正己烷层,该步骤重复3次。
净化:将下层水相溶液上样于 CleanertMCPD 固相萃取柱中,待液体完全进入硅藻土填料后,静置平衡 10 min。再用 15 mL 乙酸乙酯-正己烷体系(体积比为11∶9)洗脱3-MCPD组分,将洗脱液接收于具盖玻璃管中,于40 ℃水浴下氮吹浓缩至近干,用2 mL异辛烷溶解残渣。
衍生化:向净化后的溶液中加入 50 μL HFBI衍生试剂,旋紧盖子并涡旋混匀,置于 70 ℃恒温干燥箱中反应 20 min。衍生化完成后,取出冷却至室温,加 3 mL 水并涡旋振荡 30 s后静置分层,弃去下层水相,重复水洗 3 次后取上层有机相,经无水硫酸钠除水后装入进样瓶中,待GC-MS分析。
1.3.2 GC-MS检测
色谱条件:DB-5MS毛细管柱(30 m×0.25 mm×0.25 μm);进样口温度280 ℃;柱始温80 ℃ ,保持1 min,再以10 ℃/min升温至190 ℃,以40 ℃/min升至300 ℃,保持5 min;载气:高纯氮,流速3.0mL/min;进样方式:不分流进样;进样体积:1 μL。
质谱条件:EI+,电子倍增器增益:+250 V,灯丝电流:100 μA;阱温度:230 ℃;传输线温度:250 ℃;歧盒温度:50 ℃;溶剂延迟:3 min;质谱采集时间:3~20 min;扫描质量范围:m/z90~300; 扫描速率:0.65 s/次。
2 结果与讨论
2.1 样品制备条件的优化
水解步骤主体参照国家标准GB 5009.191—2016《食品安全国家标准 食品中氯丙醇及其脂肪酸酯含量的测定》,但在以下3个方面进行调整:(1)使用冰乙酸与20%氯化钠混合溶液(体积比1∶30)中和溶液中过量的甲醇-甲醇钠溶液。一方面降低因先加冰乙酸再加氯化钠溶液复杂操作引起的误差;另一方面3-MCPD酯的甲醇/甲醇钠溶液水解是一个快速的过程,加入乙酸与20%氯化钠混合溶液可以快速终止反应,避免碱过量造成的3-MCPD分解。(2)洗脱时,采用乙酸乙酯-正己烷体系(体积比为11∶9)作为洗脱液。实验中发现在乙酸乙酯中添加一定比例的正己烷能使之更好地匹配3-MCPD的极性,以减少非目标物质的干扰。试验结果表明乙酸乙酯-正己烷体积比为11∶9 时的回收率最高。(3)复溶时,国家标准中使用的溶液是正己烷,但正己烷在70 ℃高温的衍生化步骤中极易挥发,导致定容体积不准,因此改用挥发性较弱的异辛烷进行复溶。
2.2 3-MCPD和d5-3-MCPD的HFBI衍生物的定性分析
采用间接法检测分析3-MCPD酯,最终是通过将3-MCPD酯碱水解成3-MCPD后,再经过HFBI衍生化进行GC-MS检测分析。在3-MCPD衍生物的质谱图中(图1),m/z169 [C3F7]-、197[C3F7CO]-、225[C3F7CO2C]-离子碎片的相对丰度较大,但其均为七氟丁酰基的特征离子,并不是3-MCPD衍生物的特征离子。本文采用代表性的3-MCPD衍生物的特征离子作为定性、定量离子,如
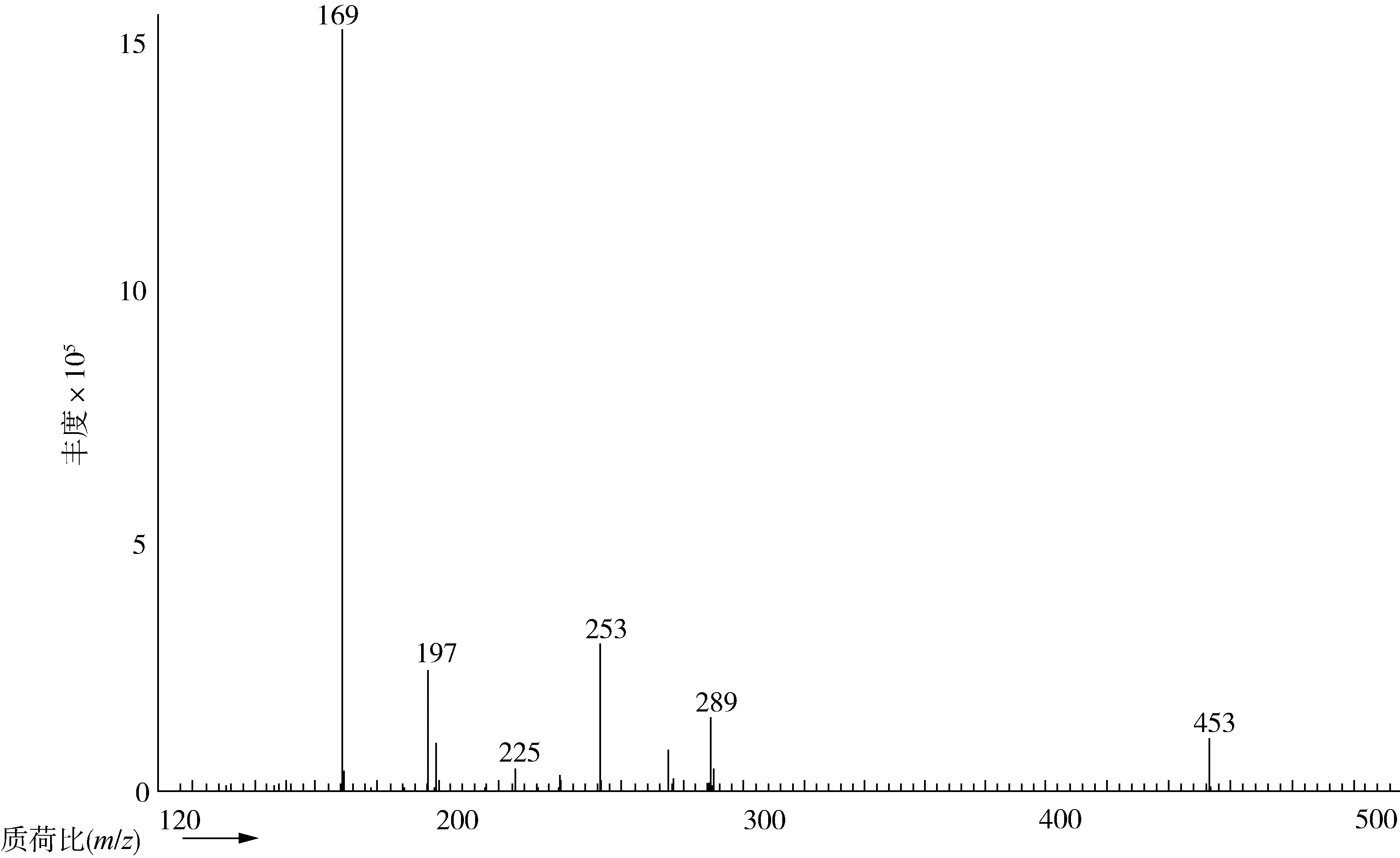
图1 3-MCPD衍生物的质谱图
表1所示。其中,m/z=253是3-MCPD的HFBI衍生物[M-C3F7CO2-HCl]+碎片离子的质荷比,m/z277(275)是[M-C3F7CO2CH2]+碎片离子的质荷比,m/z289(291)是[M-C3F7CO2]+的碎片离子质荷比,m/z453是[M-CH2Cl]+碎片离子的质荷比。相应的d5-3-MCPD的衍生物的对应离子碎片质荷比为m/z=257、278(280)、294(291)、456[12]。

表1 3-氯丙醇及其内标物衍生产物的特征离子
注:下划线离子为定量离子,其余为定性离子。
2.3 方法学评价
2.3.1 线性范围、检出限、定量限
准确移取一定量的3-MCPD以及d5-3-MCPD标准溶液于具盖试管中,用正己烷定容至2 mL,配制成相当于样品中3-MCPD含量为0.5、1.0、1.5、2.0、2.5 mg/L的一系列标准溶液。利用梯度浓度的3-MCPD标准品建立标准曲线结果如图2所示。以3-MCPD以及d5-3-MCPD衍生物的定量离子峰面积之比为纵坐标,3-MCPD质量浓度为横坐标进行线性回归。得到的线性方程为Y=0.289 9X+0.045 2,R2=0.988 5。以信噪比(S/N)为3和10时对应的加标水平分别作为方法的检测限和定量限。此方法在0.5~2.5 mg/L范围内线性相关性良好,检出限为0.025 5 mg/kg,定量限为0.093 3 mg/kg。满足油脂中3-MCPD酯的分析检测。
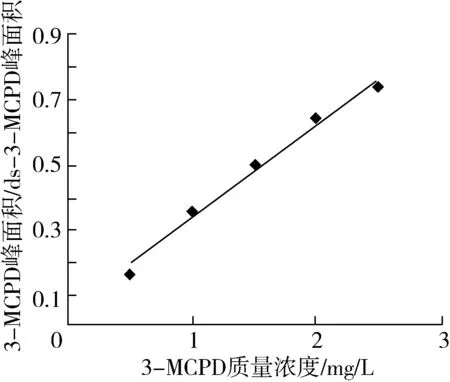
图2 3-MCPD标准曲线
2.3.2 加标回收率
取15份胡麻油样品,其中3份空白对照,其余样品中分别加入0.50、1.00、2.00、2.50 mg/kg 3-MCPD棕榈酸二酯标准液,每组做3个平行样,经过样品前处理后检测3-MCPD酯含量,计算相应的加标回收率和RSD,如表2所示。结果表明3-MCPD
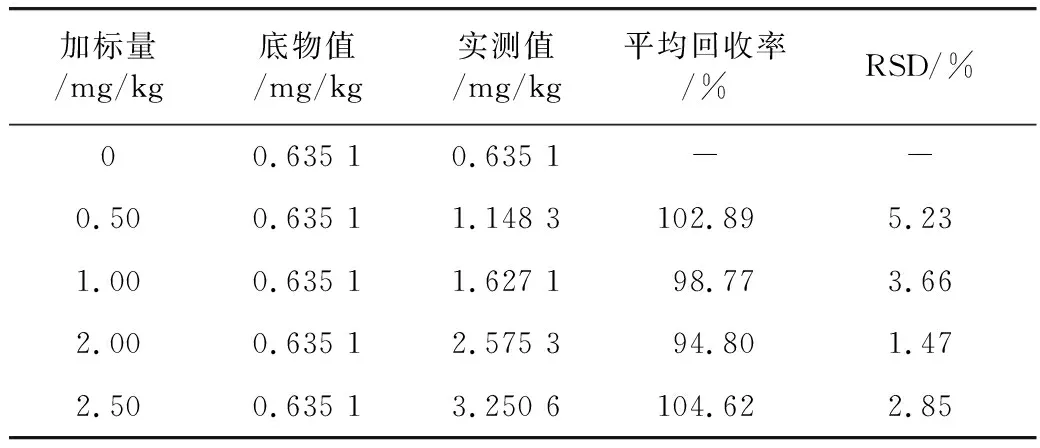
表2 加标回收率实验(n=3)
脂肪酸酯的平均加标回收率在94.80%~104.62%之间,RSD 1.47%~5.23%。表明本研究所建立的方法的准确度和精密度较高,适用于食用植物油中3-MCPD酯含量的测定。
2.3.3 仪器精密度
选取3个浓度分别为0.50、1.00、2.00 mg/kg的3-MCPD标品衍生后进行GC-MS检测,平行进样3次,计算测定值的RSD。结果如表3所示,3个浓度的3-MCPD标品的RSD值在1.83%~3.51%,表明仪器的性能良好,可以进行相关的实验。
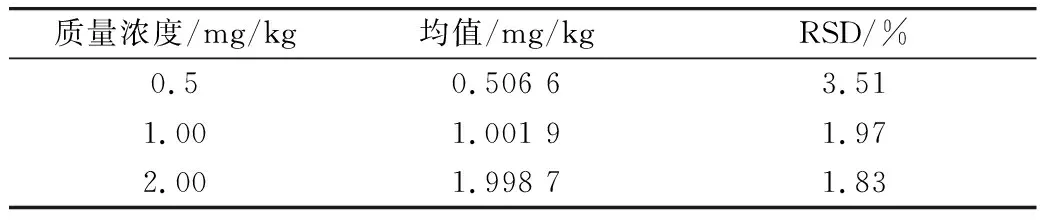
表3 仪器精密度(n=3)
2.4 新疆几种市售食用植物油中3-MCPD酯的含量
检测新疆产的几种市售食用植物油,每种油脂做3个平行样。结果取平均值如表4所示。

表4 新疆几种食用植物油中3-MCPD酯的测定结果
由表4可以看出,新疆市售的几种食用植物油中均检测出浓度不等的3-MCPD酯。在新的国家食品安全标准GB 2762—2017《食品中污染物痕量》中对于液态调味品中3-MCPD的限量为0.4 mg/kg,固态调味品中3-MCPD的限量为1.0 mg/kg。借此标准,可以看到在新疆的几种食用植物油中,菜籽油、亚麻籽油、番茄籽油、花生油以及葡萄籽油受3-MCPD酯的污染较严重,含量都高于0.4 mg/kg。其中葡萄籽油、菜籽油中3-MCPD酯的含量在1.0 mg/kg以上。而核桃油、大豆油、红花籽油中3-MCPD酯的含量都低于0.4 mg/kg。
造成食用植物油中3-MCPD酯的含量的差别一方面原因与其自身的组成成分有关。不同的食用植物油中所含有的3-MCPD酯的前体物质如三酰基甘油酯、氯离子、脂肪酸等存在着差异性[13]。另一方面与油脂的精炼工艺的差异有关。食用植物油的精炼过程的四个主要步骤:脱胶、脱酸、脱色和脱臭。在精炼的时候,若采用磷酸进行脱胶,可能会增加食用植物油中氯离子的含量,导致3-MCPD酯含量的增加;脱色白土的种类和酸性强弱也会影响食用植物油中3-MCPD的生成。目前已初步证实,精炼过程中的脱臭工序是3-MCPD酯生成的主要环节[14]。脱臭工序是在高温加热条件下去除不需要的低沸点组分。在这过程中,植物油中的氯离子与酰基甘油酯等 3-MCPD酯的前体物质通过SN1取代反应生成 3-MCPD 酯。因此,在脱臭工序前尽可能除去3-MCPD酯的前体物质可以有效的预防3-MCPD酯的生成[15-16]。或者通过适当的调整脱臭工序,比如采取分步脱臭。第一步进行低温长时间水蒸气蒸馏去除大部分不需要的易挥发组分,第二步高温短时间水蒸气蒸馏去除剩余少量高沸点组分。缩短高温加热时间,也可以有效的降低油脂中3-MCPD酯的形成。
3 结论
3.1 本实验通过甲醇钠-甲醇碱水解,固相萃取柱净化,HFBI衍生结合GC-MS方法测定食用植物油中3-MCPD酯含量。该方法在0.50~2.50 mg/L范围内线性关系良好,检出限为0.025 5 mg/kg,定量限为0.093 3 mg/kg。3-MCPD酯的平均加标回收率在94.80%~104.62%之间,RSD 1.47%~5.23%。本方法灵敏度高重复性好,能够很好的适用于食用植物油中3-MCPD酯的检测。
3.2 采用了该方法测定的新疆几种市售食用植物油中的中3-MCPD酯的含量。在新疆市售的几种食用植物油中均检测出浓度不等的3-MCPD酯。其中菜籽油、亚麻籽油、番茄籽油、花生油以及葡萄籽油受3-MCPD酯的污染较严重,3-MCPD酯的含量都高于0.4 mg/kg。而核桃油、大豆油、红花籽油中3-MCPD酯的含量都低于0.4 mg/kg,达到标准。
