汉族人群青少年特发性脊柱侧凸家系中存在复合型杂合DOCK9基因突变
2018-12-29张泽瀚孟怡辰周许辉
张泽瀚,吉 喆,姜 横,林 涛,孟怡辰*,周许辉*
1.海军军医大学附属长征医院骨科,上海 200003
2.海军军医大学基础医学院学员五队,上海 200433
3.新疆维吾尔自治区人民医院骨科中心,新疆维吾尔自治区 831000
青少年特发性脊柱侧凸(AIS)是指冠状位弯曲>10°并伴有椎体旋转的一种脊柱三维畸形[1]。AIS的病因仍然未知,双胞胎研究和家族聚集研究显示遗传作用在AIS的发生过程中起到了重要作用[2-4]。但截至目前,只有AKAP2基因和POC5基因被证实可以导致AIS[5-6]。此外,几项全基因组关联研究已经确定GPR126、CHL1和LBX1基因中的单核苷酸多态性(SNP)位点与AIS发生相关[7-8]。这些研究结果均表明AIS具有遗传异质性。本研究试图通过全基因组测序的方法来探索中国汉族AIS患者中呈复合杂合隐性遗传的致病基因。
1 资料与方法
1.1 研究对象
招募1个汉族AIS家系。家系的先证者在15岁时被确诊为AIS,她的双胞胎妹妹表现出与先证者相似的侧凸类型,其他家庭成员(父母)没有明显的脊柱侧凸病史。所有家庭成员均拍摄全脊柱正侧位X线片。由2名经验丰富的矫形外科医师独立进行体格检查和病史回顾。AIS定义为脊柱冠状面侧凸曲度>10°并伴有椎体旋转,无先天性畸形。本研究经海军军医大学附属长征医院伦理委员会批准,参与研究的家系成员均于研究前签署了书面知情同意书。
1.2 检测方法
使用血液DNA提取试剂盒(QIAamp DNA Blood Mini Kit,德国Qiagen公司)从受试者(先证者及其父母)外周血中提取DNA样品,储存于-20℃冰箱备用。用1%琼脂糖凝胶电泳评估DNA的完整性并使用Applied Biosystem SOLiD 4.0系统进行全基因组测序(WGS),依据标准SOLiD分析工作流程对主要测序数据进行分析,使用SOAPaligner程序对原始序列读数并与参考基因组(hg19)进行比对。选择非同义(NS)变体、剪接(SS)变体以及插入或缺失(indel)变体3种可能更具致病性的变体类型研究潜在致病突变。调用SNP对候选基因进行优先排序分析,比对读数去除dbSNP、HapMap、1000 Genomes、ESP6500、ExAC以及中国内地外显子数据库(1 500个中国汉族个体)中存在的序列变异[9]。使用SIFT(sift.bii.a-star.edu.sg/)、Polyphen-2(genetics.bwh.harvard.edu/pph2/)和MutationTaster(www.mutationtaster.org/)预测有破坏性的单核苷酸变异(SNV)。
1.3 验证方法
使用Primer 5程序(https://frodo.wi.mit.edu/)设计PCR引物,对基因组DNA进行PCR扩增,在ABI 3130 DNA分析仪上进行双向DNA Sanger测序验证所有发现的突变。
2 结 果
本研究纳入了了一个二代家庭,先证者主胸弯Cobb角为46°,于2015年在长征医院接受了矫形手术(图1a,b),其妹妹具有相似的侧凸,Cobb角21°,未接受治疗,密切临床随访中(图1c,d),其他家庭成员(父母)没有脊柱畸形表现,AIS在这个家庭中表现为隐性遗传或新发性状(de novo)。WGS在鉴定常染色体隐性遗传方面已被证明非常成功。本研究准备了先证者及其父母3个人的基因组文库,获得了平均深度为38.15×、人均830.87 Mb的序列,测序的数据详见表1。
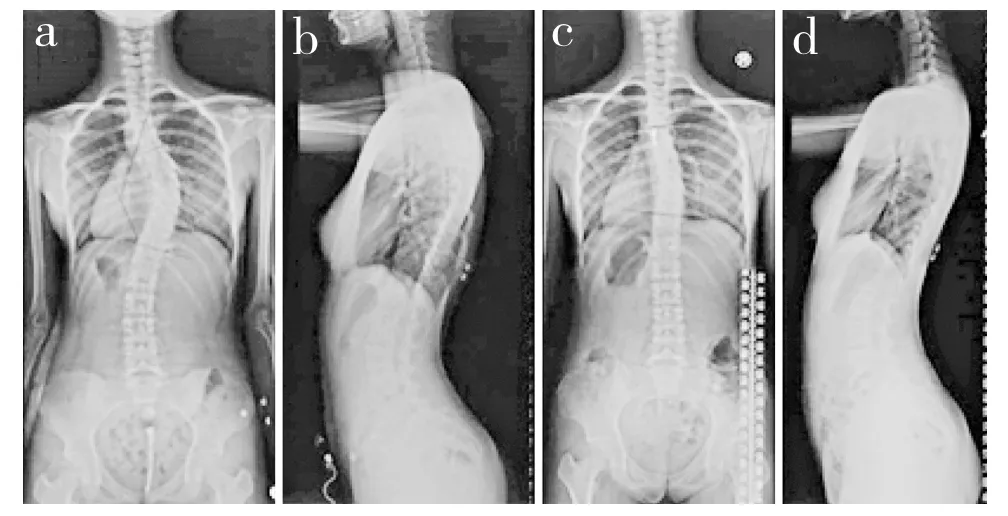
图1 全脊柱X线片Fig. 1 Whole spine roentgenographs
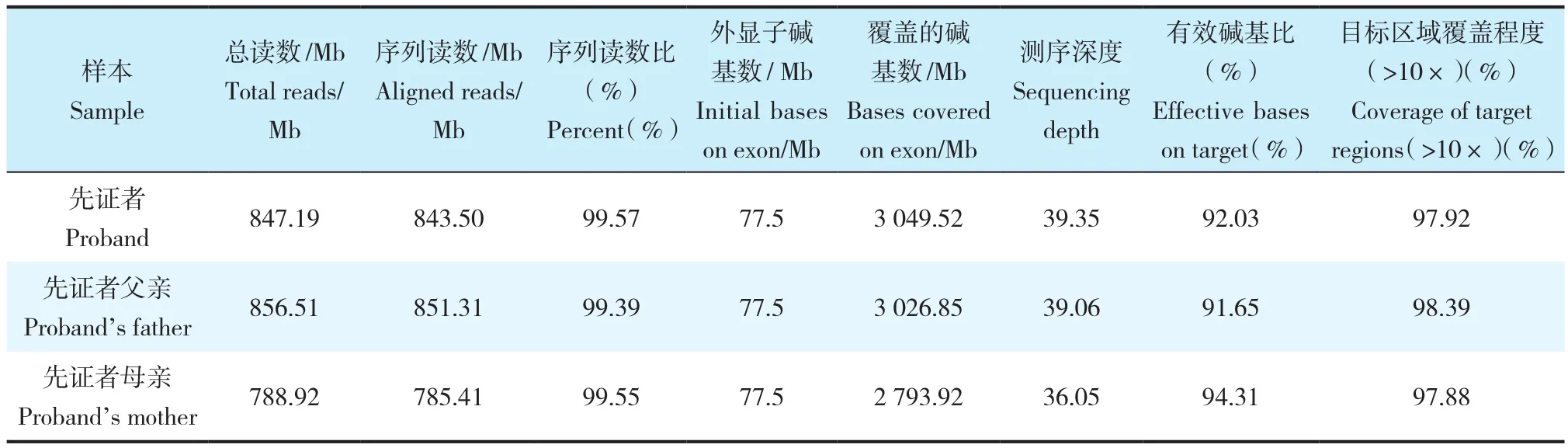
表1 WGS数据Tab. 1 Data of WGS
将平均99.50%原始序列读数与参考基因组(hg19)进行比对,分析2种模式(隐性遗传或新发性状)可能性,其中新发性状所有识别的突变都呈假阳性,只有复合杂合隐性遗传模式符合该家系遗传模式。先证者存在NS/SS/indel变体,但其父母不存在变体。排除所有数据库中频率>0.01%的突变,考虑到大多数致病变体可能影响高度保守的序列和/或被预测为有害,使用SIFT、PolyPhen-2和MutationTaster的预测程序得分来预测变体的功能影响,得到8个基因,其中7个基因在功能上无关而排除(表2),只有DOCK9基因的2个突变保留作为候选变体:外显子30中的错义突变c.3259T>C(p.F1087L,NM_001130050)和外显子22中的错义突变 c.2465A>G(p.Y822C,NM_001130050)。2种突变均通过Sanger测序证实(图2),并且突变位点在不同物种氨基酸水平上高度保守(图3)。在该家系中,先证者DOCK9基因中存在该复合型杂合突变,经验证,其妹妹的DOCK9基因中也存在该复合型杂合突变,而未发现既往报道的基因突变(POC5、AKAP2)。其母携带错义突变p.F1087L,其父携带错义突变p.Y822C,据此推测DOCK9突变基因很可能是AIS的发生原因。
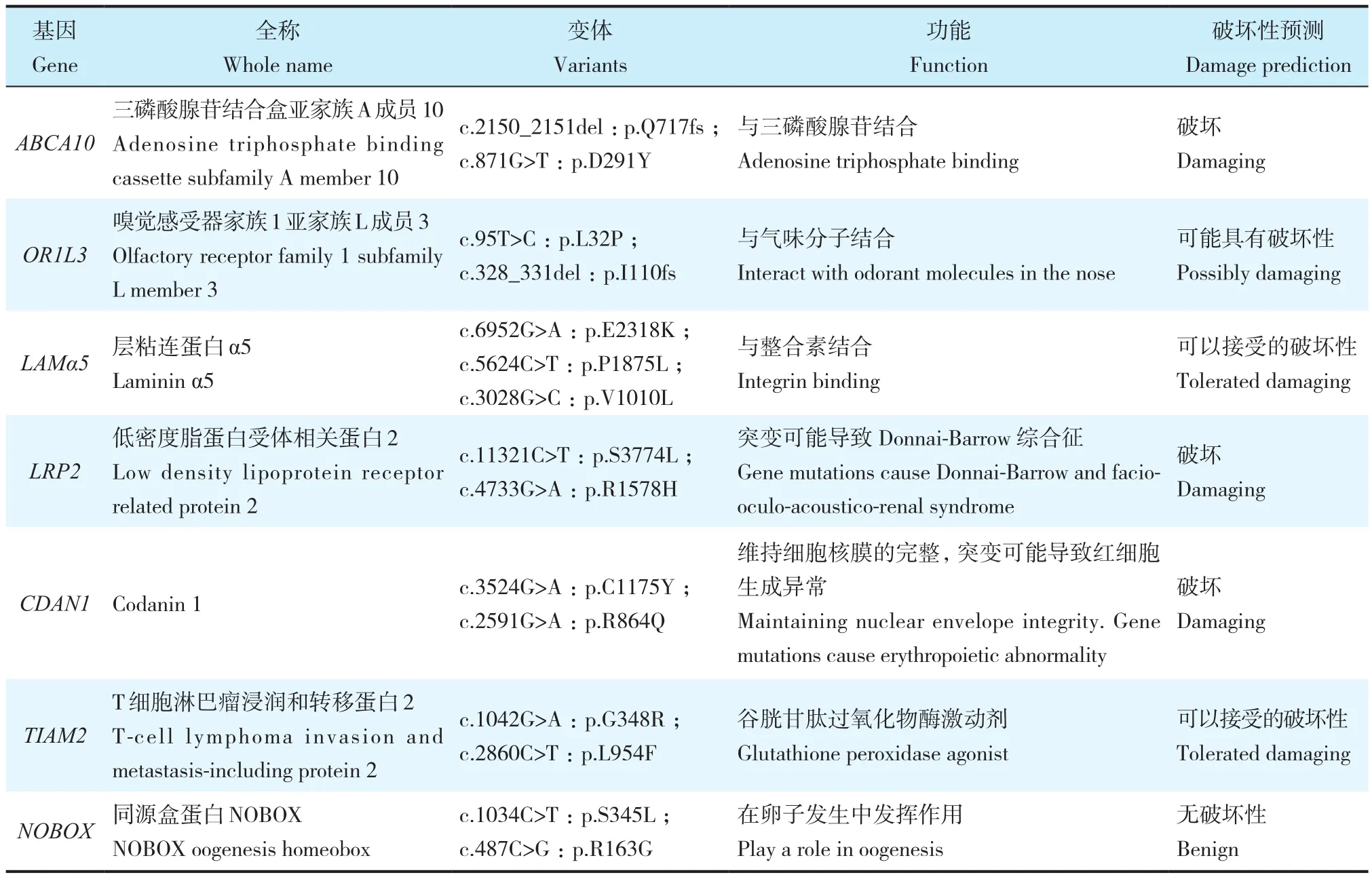
表2 WGS排除基因Tab. 2 Excluded gene of WGS

图2 Sanger测序验证DOCK9突变Fig. 2 Sanger sequencing for verifying DOCK9 mutation

图3 突变位点在不同物种之间具有高度保守性Fig. 3 Mutation sites with highly evolutionary conservation between multiple species
3 讨 论
AIS的病因尚不明确,但是既往的流行病学研究显示,直系亲属中存在AIS患者,AIS发生风险较高,而且在双胞胎研究中,同卵双胞胎同时出现脊柱侧凸表型的比例(73%)明显高于异卵双胞胎(36%)[10],表明遗传因素在AIS发生过程中起到关键作用。近年研究已证实AKAP2、POC5为AIS的致病基因[5-6],GPR126、CHL1和LBX1基因也与AIS发生相关[7-8]。但是这些基因只能解释少部分AIS患者的发病机制,还有大部分患者的发病原因不能用已经报道的突变基因解释。
本研究对1个汉族AIS双胞胎家系进行了探究,通过测序发现,位于DOCK9基因30号外显子的错义突变c.3259T>C(p.F1087L)和位于DOCK9基因22号外显子的错义突变c.2465A>G(p.Y822C)可能是该家系AIS发生的原因。该家系中的2名患者在DOCK9基因中都存在该复合型杂合突变,先证者父亲仅携带p.Y822C突变,其母仅携带p.F1087L突变。DOCK9基因位于13号染色体长臂32.3区域,有61个外显子。其编码的DOCK9蛋白属于Rho鸟嘌呤核苷酸交换因子,通过将鸟苷二磷酸转化为鸟苷三磷酸而激活Rho鸟苷三磷酸酶,进一步调控细胞的迁移、极性,细胞骨架的组装以及细胞凋亡[11]。DOCK9可特异性地与cdc42结合形成复合体。Choi等[12]研究发现,cdc42基因敲除的斑马鱼模型出现脊柱侧凸的表型;而对斑马鱼进行解剖发现,其神经管内的纤毛发育异常。纤毛是一类传递化学及机械信号的细胞器,在人类组织中(除血细胞)广泛存在。Grimes等[13]研究发现,ptk7基因突变的斑马鱼室管膜细胞纤毛发育和脑脊液循环出现障碍,表现出类似于AIS的表型,证实ptk7基因突变斑马鱼是研究AIS的理想动物模型,也为纤毛功能障碍在AIS的发生中发挥重要作用提供了证据。此外,Baek等[14]已经证明,Tuba蛋白的缺乏可导致斑马鱼纤毛发育障碍,出现脊柱畸形的表型,而Tuba与DOCK9具有相同的功能。Oliazadeh等[15]研究发现,AIS患者成骨细胞纤毛长度较正常人的成骨细胞长,而纤毛的长度与信号转导的强弱有关;因此,也认为纤毛的功能异常在AIS的发生过程中起关键作用。因此,本研究组推测DOCK9-cdc42途径可能在纤毛发育中起关键作用,而其通路异常可能导致脊柱弯曲,出现AIS表型。下一步将继续围绕DOCK9在纤毛发育中的具体作用进行深入的机制研究。
总之,本研究首次发现AIS患者家族中DOCK9基因突变的存在,并且以复合型杂合隐性方式遗传给后代。今后需要构建动物模型对DOCK9在纤毛发育中的具体机制进行深入探讨,以解释DOCK9基因突变在AIS发生机制中的作用。
