C H F=C H C l分子的特性研究
2018-12-28梁冬梅孙光宇
梁冬梅 荆 涛 * 孙光宇
(1.凯里学院理学院,贵州 凯里 556011;2.贵州师范学院物理与电子工程学院,贵州 贵阳 550018)
0 引言
近年来的实验和理论研究发现, 二卤代乙烯类化合物顺式构型的稳定性明显优于反式构型的稳定性[1-4]。并且,樊晓伟等[5]对化合物二氟代乙烯使用不同的基组进行计算发现, 使用小基组计算的总能量和轨道能量并不收敛, 甚至会给出错误的能级排序, 而使用较大基组计算的数值更接近于实验值。 因此, 为了给出正确的计算结果,本文使用与文献[5]相同的基组进行计算。
1 理论和计算方法
外电场存在时,哈密顿量H 可用下式求出,H=H0+Hint, (1)
上式中Hint的含义为, 当分子处在偶极近似条件下,外电场和分子体系之间的相互作用。 其表达式为,Hint=-μ·F (2)
在(2)式中,变量μ 表示偶极矩。
若哈密顿量Hint为零,有H=H0, (3)
从Grozema 等[6-7]创立的模型看,有外电场存在时的激发能Eexc为,
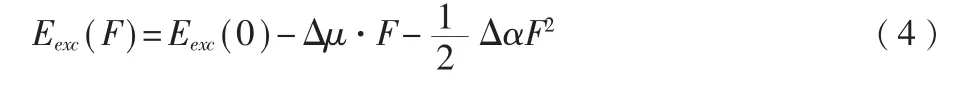
其中,在无外电场时的激发能,Eexc(F)=Eexc(0)。(5)

在上面的公式中包含了几个重要的量, 它们分别是加权因子、线强度和波数,用字母表示为gl、S、σ,加权因子的大小为1,线强度的单位为e2a2
0。
按照CHF=CHF 分子的标准坐标, 在B3LYP/6-311++G(3df, 3pd)基组的基础上对该分子的基态结构进行优化, 在此基础上对其激发态能量等物理量进行了计算。 所有计算均在Gaussian03 软件包[9]进行。
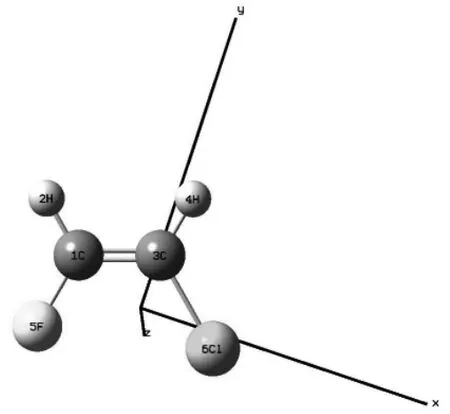
图1 CHF=CHCl 分子的基态结构
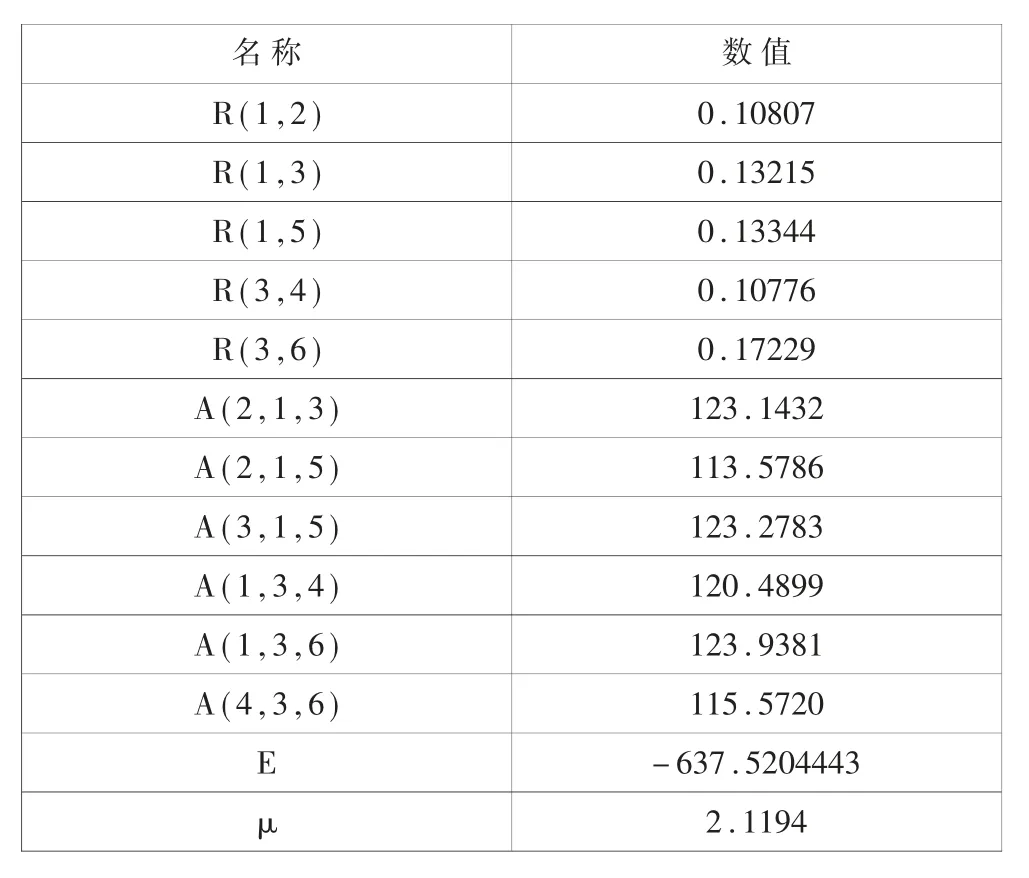
表1 优化的CHF=CHCl 分子基态键长(R/nm)、键角(A/deg)、总能量(E/a.u.)和偶极矩(μ/debye)
2 计算结果与讨论
2.1 CHF=CHCl 分子的基态稳定构型
CHF=CHCl 分子的结构如图1 所示。采用密度泛函理论(DFT)中的B3LYP,以6-311++G(3df,3pd)作为基函数,对其进行结构优化,得到该分子的基态键长、键角和总能,如表1 所示。
从表1 可以看出,分子的键长在0.10776~0.17229 nm之间变化,键角在113.5786~123.2783deg 之间变化,基态总能量为-637.5204443 a.u.,偶极矩为2.1194。
2.2 CHF=CHCl 分子的激发特性
在得到CHF=CHCl 分子稳定构型的基础上, 采用CIS-B3LYP/6-311++G(3df, 3pd)方法研究了该分子的前8 个激发态的激发能E、激发波长λ 和振子强度,如表2 所示。

表2 分子CHF=CHCl 的激发态能量、激发波长和振子强度
通过表2 可以得出以下规律, 分子从基态跃迁到前8 个激发态, 激发态能量不断增大, 激发态波长不断减少。 如图2 所示, 激发能从6.8258eV 增加到9.0568eV,而波长从181.64nm 下降到136.90nm。 振子强度就是振子数,从表2 可以看出,在从基态跃迁到激发态的过程中,振子强度并不为零,但大小不同。特别是从基态跃迁到第三个激发态, 振子强度f 达到了0.5218,有明显的电子跃迁发生, 而从基态跃迁到第七个激发态,振子强度f 为0.0325,振子强度比较小,在实验上即使有电子跃迁发生,也非常微弱,基本上观察不到。 其它从基态跃迁到此激发态(n=1,2,4,5,6,8),振子强度非常小,在实验上观察不到跃迁发生。
3 结论
本文采用B3LYP/6-311++G(3df,3pd)方法得到了CHF=CHCl 分子的基态稳定构型, 讨论了分子的总能量、偶极矩和激发特性。 结论如下:
(1) 分 子 的 键 长 在0.10776 ~0.17229nm 之 间 变 化,键角在113.5786 ~123.2783deg 之间变化, 基态总能量为-637.5204443 a.u.,偶极矩为2.1194。
(2) 采用CIS-DFT 方法研究了分子的激发特性。分子的激发波长随激发能的增大逐渐减小, 变化的快慢与激发能的变化快慢相关, 但并没有在可见光区出现。 振子强度的大小不受激发能的影响, 电子跃迁的情况较为复杂。 从基态跃迁到第三激发态, 可以观察到明显的电子跃迁, 其它情况的振子强度较小, 基本上观察不到跃迁现象发生。
