富马酸替诺福韦二吡呋酯中6种有关物质高效液相色谱测定方法改进
2018-12-25杨智慧
程 诚,杨智慧
(江苏省连云港市食品药品检验检测中心,江苏 连云港 222000)
富马酸替诺福韦二吡呋酯是美国Gilead Sciences公司研制的新型核苷酸逆转录酶抑制剂,临床主要用于治疗人类免疫缺陷病毒(HIV)感染[1-4];同时可通过干扰乙型病毒型肝炎(简称乙肝)病毒DNA聚合酶的功能而抑制乙肝病毒的复制[5],降低血清及肝组织内的病毒载量。目前关于该药的质量分析已有研究报道[6-12],有关物质一般采用高效液相色谱(HPLC)法进行检测[13-15]。本研究中对该药生产工艺中可能存在的 6种有关物质对照品进行收集和分析[16-18],同时建立了采用HPLC法梯度洗脱测定该药原料药中6种有关物质的方法。现报道如下。
1 仪器与试药
1.1 仪器
U3000型高效液相色谱仪(美国戴安公司);XS105型电子天平(瑞士梅特勒-托利多公司)。
1.2 试药
富马酸替诺福韦二吡呋酯对照品(批号为G0M335,USP,纯 度 为 98.4% );杂 质 C(批 号 为R044C0,USP,纯 度 为 100.0% );杂 质 D(批 号 为10M090,USP,纯度为 99.9% );杂质 E(批号为 1937 -020A7,TLC,纯度为 100.0% );杂质 F(批号为 1508 -099A3,TLC,纯 度 为 100.0% );杂 质 G(批 号 为R038N0,USP,纯度为 98.0% );富马酸对照品(批号为111541-201102,中国食品药品检定研究院,纯度为99.4%);富马酸替诺福韦二吡呋酯样品(石家庄龙泽制药股份有限公司,批号分别为180101,180102,280201,纯度均为99.8%);甲醇、叔丁醇为色谱纯,其余试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Waters Symmery C18柱(250 mm ×4.6 mm,5 μm,美国 Waters公司);流速:1.0 mL /min;柱温:30 ℃;检测波长:260 nm;流动相:以甲醇 -0.05 mol/L磷酸氢二钠溶液(用磷酸调节 pH 至 5.0)-叔丁醇(15 ∶22 ∶3)为流动相 A,以甲醇 -0.05 mol/L磷酸氢二钠溶液(用磷酸调节 pH 至 5.0)-叔丁醇(30 ∶7 ∶3)为流动相 B,按表1条件进行梯度洗脱。
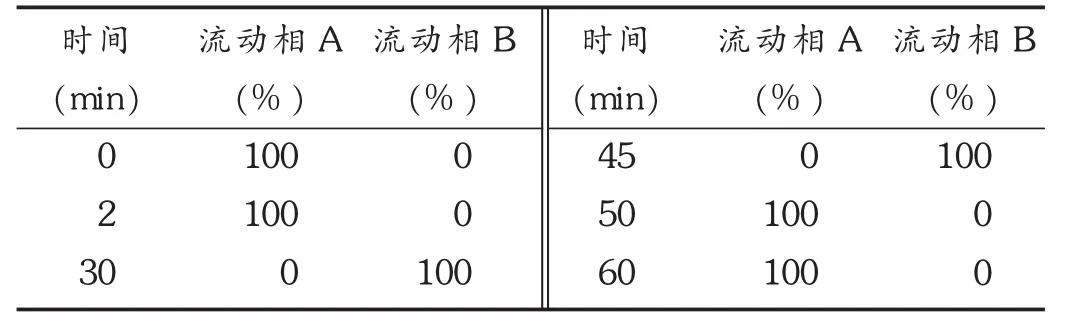
表1 梯度洗脱条件
2.2 溶液制备
杂质对照品溶液:称取富马酸、杂质 E、杂质 F、杂质G、杂质C、杂质D对照品适量,精密称定,加入流动相A溶解并稀释成各成分质量浓度约为75 μg/mL的溶液,作为杂质对照品贮备液;取该贮备液适量,用流动相A稀释成各成分质量浓度约为0.75 μg/mL的溶液,作为杂质对照品溶液。
供试品溶液:取富马酸替诺福韦二吡呋酯样品,精密称定,加流动相A溶解并稀释成质量浓度为0.5 g/L的溶液,作为供试品溶液。
2.3 方法学考察
系统适用性试验:称取富马酸、杂质 E、杂质 F、杂质G、杂质C、杂质D与富马酸替诺福韦二吡呋酯对照品适量,精密称定,加入流动相A溶解并稀释成各成分质量浓度约为 0.75 μg/mL的溶液,进样测定。系统适用性试验色谱图见图1。
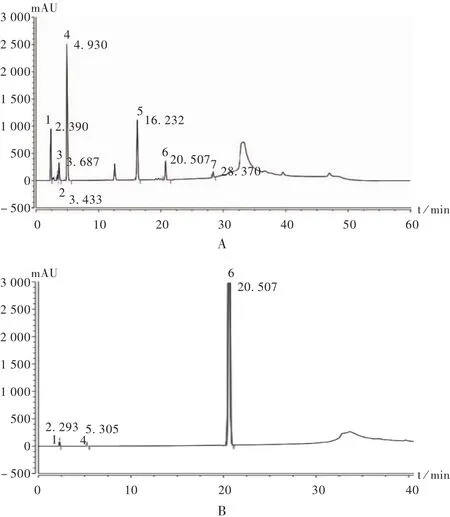
图1 系统适用性试验色谱图
专属性试验:为考察本色谱条件下主峰与各杂质峰的分离情况,对该制剂进行了酸(加1 mol/L盐酸溶液1 mL)、碱(加 1 mol/L 氢氧化钠溶液 1 mL)、高温(105℃)、氧化(加30%过氧化氢1 mL)与光照(置光照度为4 500 lx环境)破坏性试验,分别放置6 h后,按供试品溶液制备方法配制降解产物溶液后进样测定,记录色谱图(见图2)。结果表明,本品在高温、光照条件下较稳定,氧化、酸、碱条件下稍有降解,主成分峰能与降解产物峰良好分离,方法专属性良好,可用于该制剂中有关物质的检测。
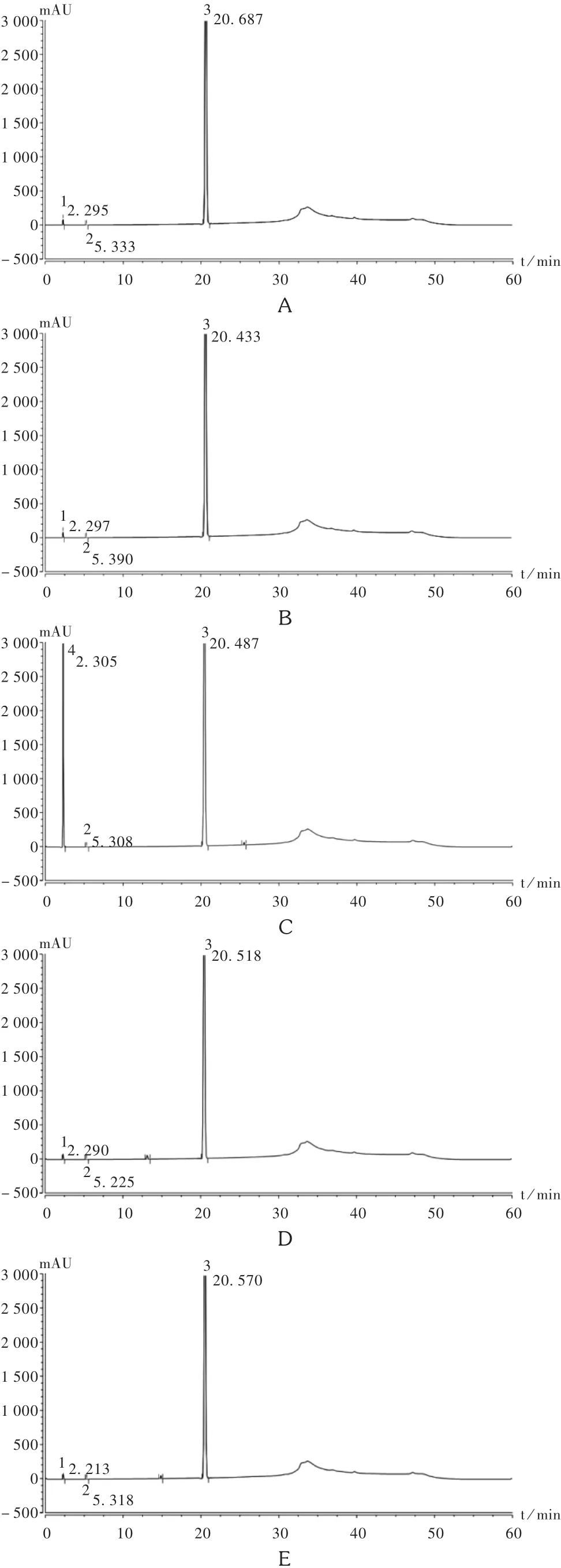
图2 专属性试验色谱图
线性关系考察:取各杂质对照品适量,分别配制成质量浓度为 0.375,0.750,3.750,7.500,20.000 μg /mL的系列溶液,各精密量取 10 μL,按 2.1项下色谱条件检测,记录色谱图,以峰面积(Y)为纵坐标、进样质量浓度(X,μg/mL)为横坐标进行线性回归。结果见表 2。
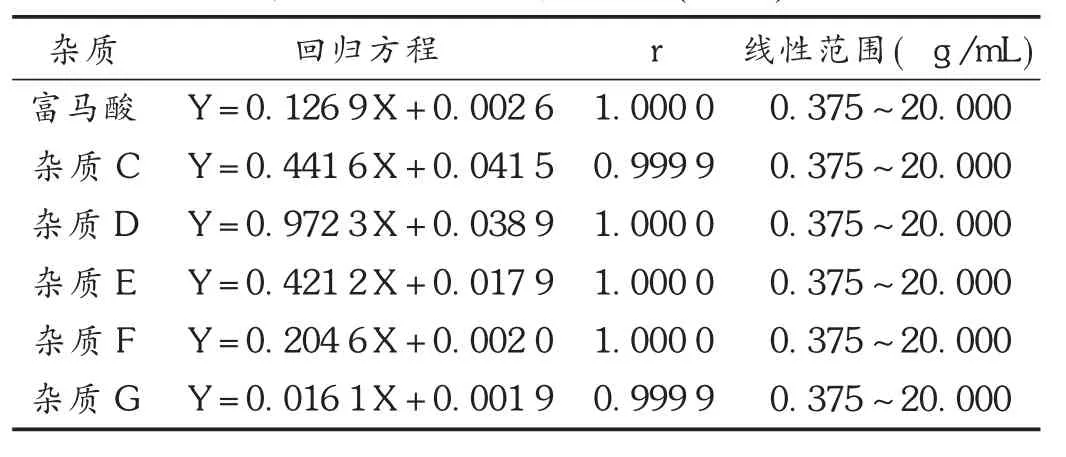
表2 线性关系考察结果(n=5)
检出限和定量限测定:取“线性关系考察”项下最低质量浓度的对照品溶液,逐级稀释,按2.1项下色谱条件进行检测。信噪比(S/N)≥3时,富马酸、杂质 D、杂质 E、杂质F的检出限为7.5 ng/mL,杂质C和杂质G的检出限为0.15 μg/mL;S/N≥10 时,富马酸、杂质D、杂质 E、杂质F的定量限为15 ng/mL,杂质C和杂质G的定量限为0.375 μg /mL。
重复性试验:依法配制供试品溶液6份,按2.1项下色谱条件检测,计算6份供试品溶液中富马酸替诺福韦二吡呋酯主峰面积与称样量的比值(A/m),结果的RSD为0.79%(n=6),表明方法重复性良好。
精密度试验:依法配制供试品溶液1份,连续进样6次,记录富马酸替诺福韦二吡呋酯主峰面积。结果的RSD为0.46%(n=6),表明仪器精密度良好。
稳定性试验:依法配制供试品溶液,分别于0,4,8,12,24 h时按2.1项下色谱条件进样检测。结果富马酸替诺福韦二吡呋酯主峰面积的 RSD为0.63%(n=5),同时没有新杂质产生,表明供试品溶液在24 h内稳定。
回收率试验:依法配制供试品溶液,分别加入不同体积的杂质对照品贮备液,配成含各杂质质量浓度分别为 0.75,1.00,1.50 μg/mL 的溶液,各平行配制 3 份,进样检测,计算回收率。结果见表3。
2.4 样品含量测定
取3批样品,依法制备供试品溶液,按2.1项下色谱条件进行测定。试验数据见表4。可见,3批样品种富马酸和杂质E均有检出,表明建立的检测方法可有效控制富马酸替诺福韦二吡呋酯的质量。
3 讨论
3.1 杂质来源分析
分析富马酸替诺福韦二吡呋酯合成生产工艺,确定了6种可能存在的有关物质。富马酸为成盐过程的合成原料;杂质C为替诺福韦,为合成中间体;杂质D为腺嘌呤,为合成原料;杂质 E 为 9-[(R)-2-(羟基)(异丙氧基羰基)氧基]甲氧基]氧膦基]甲氧基]-丙基]腺嘌呤,是替诺福韦二吡呋酯脱去1个异丙氧基羰基氧基甲氧基的反应副产物;杂质F为替诺福韦二吡呋酯中的异丙氧基被乙氧基取代的副产物;杂质G为替诺福韦二吡呋酯脱去一个甲氧基羰基氧基的副产物。可见,有必要对这6种杂质进行质量控制。
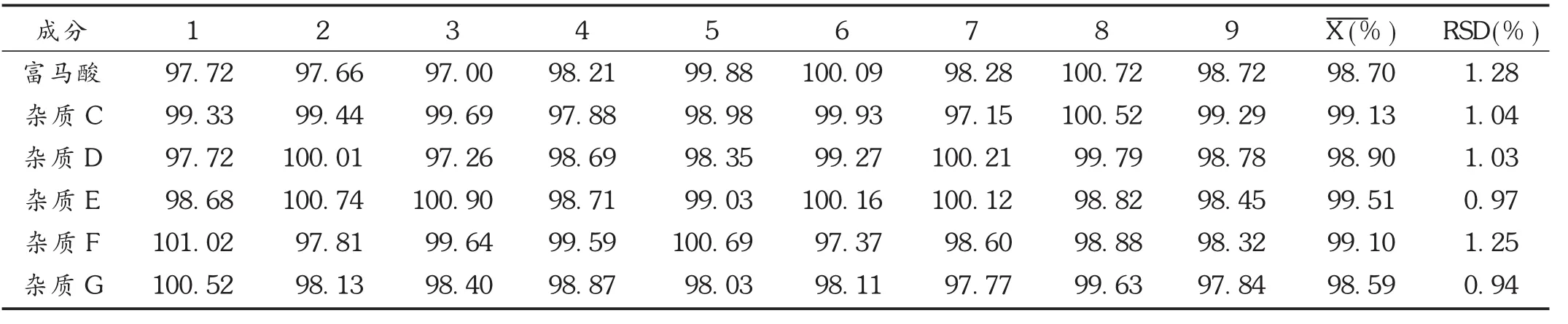
表3 各杂质回收率试验结果(%,n=9)
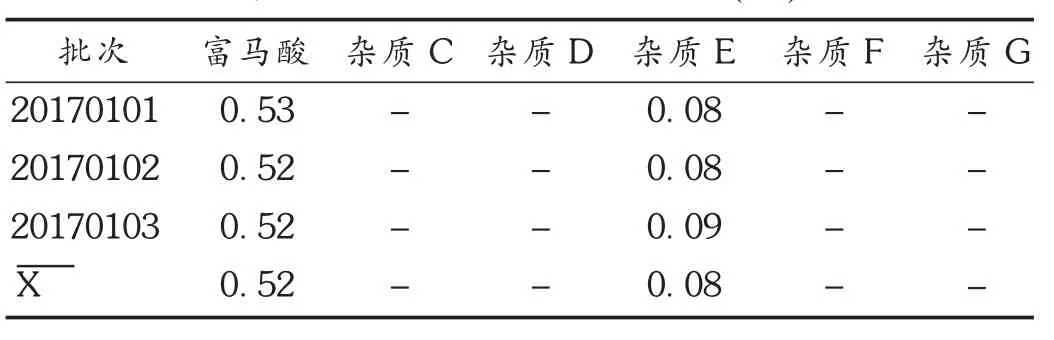
表4 3批样品含量测定结果(%)
3.2 流动相及检测波长选择
富马酸替诺福韦二吡呋酯和6种有关物质在极性较大的有机溶剂中溶解度好,同时供试品中反应起始物、中间体及副反应产物共涉及6种成分,等度洗脱不易分离且检测时间较长,最后采用反相色谱梯度洗脱进行检测,实现了良好的分离度和适当的检测时间。对富马酸替诺福韦二吡呋酯供试品和6种有关物质的溶液进行全波长紫外扫描,结果显示,检测波长为260 nm时可采用外标法有效地对该6种有关物质进行限量检测。
3.3 方法评价
本试验中建立的测定富马酸替诺福韦二吡呋酯中6种有关物质的HPLC梯度洗脱法,操作简单,专属性强,准确度高,可有效控制该制剂原料药的质量,同时也为该药相关制剂的质量控制提供了参考。
