BLM解旋酶基因的克隆、表达载体构建及表达研究
2018-11-30赵佳福许厚强宋书弦段志强陈祥
赵佳福 许厚强 宋书弦 段志强 陈祥
(1. 贵州大学高原山地动物遗传育种与繁殖教育部重点实验室 贵州大学动物科学学院,贵阳 550025;2. 贵州大学生命科学学院,贵阳550025;3. 贵州省大方县农牧局,大方 551600)
BLM解旋酶是人类RecQ解旋酶家族中的一员,具有RecQ解旋酶家族典型的结构特征,在DNA的复制、重组、转录、修复、端粒的维持等细胞代谢过程中具有重要的作用[1]。研究表明,BLM基因的突变会导致Bloom综合症的发生[2],患者遗传不稳定,并易患乳腺癌、肺癌、前列腺癌和恶性肿瘤等各种癌症[3-8],属于高风险人群。目前,BLM基因已被定位于15q26.1区带上,转录生成4.5 kb的mRNA,编码一个由1 417个氨基酸残基组成的分子量约为159 kD的蛋白[9]。已报道的资料显示,BLM解旋酶主要包含链退火结构域(Strand annealing domain,SA)、解旋酶结构域(Helicase domain)、RecQ羧 基 端 结 构 域(RecQ C-terminal domain,RQC)、解旋酶和RNA酶D羧基端结构域(Helicase and RNaseD C-terminal domain,HRDC)和核定位信号结构域(Nuclear localization signal domain,NLS),其中RecQ、RQC和HRDC位于BLM解旋酶氨基酸639-1 290位,见图1。该区域具有类似BLM全酶的活性,但缺乏羧基端的核定位功能和氨基端链退火结构域的相关功能[10-11]。目前已发表的文章大多以BLM642-1290的核心区域为研究对象,开展BLM解旋酶结构与功能的研究。本实验旨在克隆BLM解旋酶基因全长4 251 bp的CDS区域,构建BLM解旋酶全基因的重组真核表达载体和重组原核表达载体,并研究其原核细胞中的表达水平,在真核细胞中的亚细胞定位情况,以及BLM基因超表达后,对细胞核定位相关基因Ran表达水平的影响,进一步为BLM解旋酶结构与功能研究提供材料和奠定基础。

图1 人BLM解旋酶全酶结构域示意图
1 材料与方法
1.1 材料
JM109、BL21(DE3)感受态细胞、pMD19-T vector、DL10000、DL5000 DNA Marker、 限 制 性内切酶及T4 DNA ligase(宝生物),PC3细胞、pEGFP-N3载体、pET-32a(实验室储存),DNA 凝胶回收试剂盒、质粒DNA小量提取试剂盒(Axygen),RNA 提取试剂盒(Omega),无内毒素质粒提取试剂盒(QIAGEN),2×Taq PCR Master Mix、反转录试剂盒(康为),FuGENE® HD Transfection Reagent(Roche),10%胎牛血清、RPMI1640生长培养基(gibco)。抗His标签鼠单克隆抗体、HRP标记山羊抗小鼠IgG抗体(康为),抗BLM蛋白鼠单克隆抗体(Santa Cruz Biotechnology),ECL超灵敏显色试剂盒、TBSTw液封闭、彩虹Marker(碧云天)。
1.2 方法
1.2.1 引物设计 由于BLM基因较长,为提高扩增效率,采用分段扩增的方式,进行BLM全基因的克隆。根据GenBank中登录的BLM基因cDNA序列(NM_000057),采用 Primer Premier 5.0设计BLM1-1950,BLM1319-2735,BLM1-42513对引物,根据表达载体pET-32a载体图谱和BLM基因全长序列的特点,分别在B1-F、B3-R中添加KpnI、XhoI酶切位点,3对引物扩增获得的3条序列彼此间均具有部分重复序列,便于后续扩增BLM1-4251全长。此外 根 据 GenBank中 人BLM(NM_000057)、RAN(NM_006325.4)、GAPDH(NM_001289 746.1)基因的cDNA序列,设计荧光定量PCR引物,见表1。
1.2.2BLM基因的克隆 收集对数生长期的前列腺癌PC3细胞,约5×106个,采用Omega公司 RNA提取试剂盒提取细胞总RNA,反转录成cDNA,分别扩增B1、B2、B3序列,EcoR I和NcoI分别单酶切B1和B3、双酶切B2,胶回收目的片段后,T4DNA连接酶连接回收后,以回收产物为模板,采用B1-F、B3-R扩增BLM1-4251全长序列(图2);电泳检测PCR产物,胶回收目的片段,并与pMD-19-T 载体连接转化大肠杆菌JM109感受态细胞。由于目的片段较大,菌液PCR同样分为B1、B2、B3三段进行检测,最后将检测正确的重组质粒pMD-19-BLM送上海英骏生物技术公司测序鉴定。

表1 实验所使用的引物
1.2.3 重组原核和重组真核表达载体的构建 根据pET-32a(+)和pMD19-BLM载体酶切位点图谱,分别采用KpnI和XhoI限制性内切酶切割两个质粒,回收5 900 bp的pET-32a(+)载体和4 251 bp的BLM基因两条目的条带,16℃过夜连接,转化BL21感受态细胞,氨苄青霉素抗性平板筛选单克隆,挑取单克隆,摇菌并进行菌液PCR分段检测和双酶切检测,最后将PCR和双酶切鉴定均正确的pET-32a-BLM阳性菌株,以终浓度为25%的甘油进行-80℃保藏。同上,选择Hind Ⅲ和BamHⅠ两个限制性内切酶进行双酶切反应,胶回收4 700 bp的pEGFP-N3载体片段和4 251 bp的BLM基因目的片段,连接转化JM109感受态细胞,采用含卡那霉素的抗性平板进行筛选,构建重组真核表达载体pEGFP-N3-BLM。
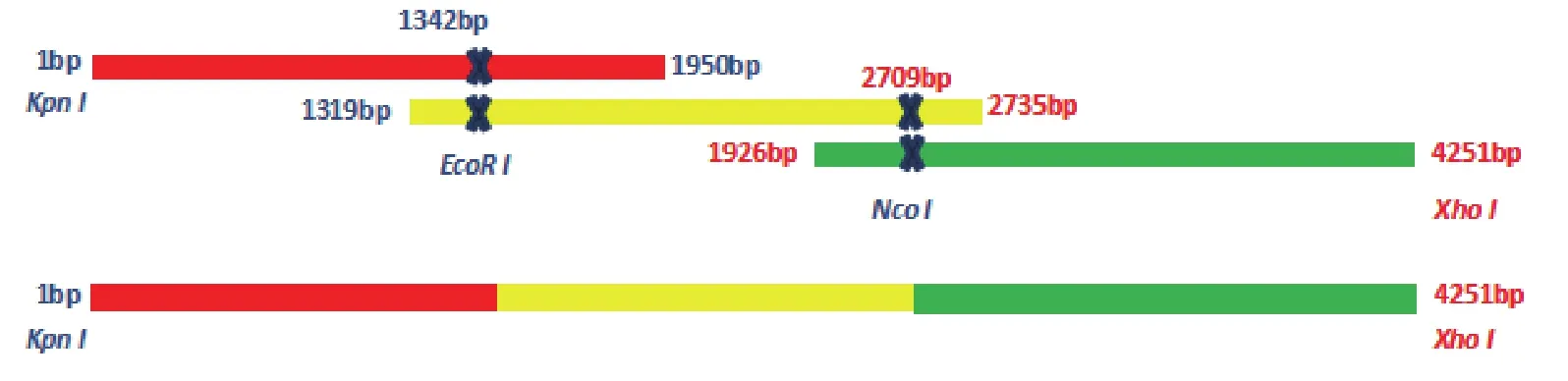
图2 BLM CDS区扩增方案示意图
1.2.4 BLM解旋酶的在大肠杆菌中的表达分析 为摸索出BLM蛋白表达的最佳培养条件,按照培养温度设为25℃和37℃两组,每组设4个IPTG 浓度梯度,终浓度分别为0.3 mmol/L、0.5 mmol/L、0.7 mmol/L和1.0 mmol/L,180 rpm条件下摇菌5 h。5 h后,按照细菌蛋白抽提试剂盒说明裂解细胞,提取总蛋白,并分离上清和沉淀,同时设含空质粒pET-32a(+)的菌株为对照组。6%分离胶进行SDS-PAGE分析,按彩虹Marker5 mL/孔、蛋白样品30 mL/孔进行上样,电压:80 V 30 min,120 V 60 min,电泳结束后120 V 2.5 h转移至PVDF膜上,TBSTw液封闭10 min,抗His标签鼠一抗4℃孵育过夜,HRP山羊抗小鼠二抗孵育1 h,DAB显色,荧光化学发光成像系统(Bio-Rad公司)对Western-blot进行结果分析。
1.2.5 BLM解旋酶在PC3细胞中的亚细胞定位分析 首先采用PSORT II Prediction在线分析软件对BLM解旋酶氨基酸序列进行分析,对其在细胞中可能的表达部位进行预测。然后将PC3细胞按照3×105/孔接种于6孔板中,37℃,5% CO2的培养箱中培养,待细胞长至80%时,按照FuGENE®HD Transfection Reagent的操作说明,将纯化后的pEGFP-N3和pEGFP-N3-BLM两个质粒分别转染PC3细胞,通过细胞转染结合荧光共定位的方法对预测结果进行验证。具体操作步骤:(1)首先弃去6孔板中培养基,用PBS清洗培养孔两次,每孔加入2 mL的含血清不含抗生素的DMEM/F-12培养基,置于37℃、5% 的CO2培养箱中培养;(2)取6支无菌PCR管,每管先加入2 µg转染质粒,再加入10 µL转染试剂,移液器轻轻混匀,再加入100 µL Opti-MEM®轻轻混匀,室温孵育30 min;(3)将转染混合物对号加入6孔板中,十字交叉法轻轻振荡混匀,继续置于5% CO2培养箱中培养。
转染细胞36 h后开始染核,具体操作步骤如下:(1)用PBS浸洗细胞3次,加入事先预冷的4%多聚甲醛固定细胞20 min;(2)PBS浸洗细胞3次,加入0.25%Triton X-100(PBS配制)室温通透5 min;以下步骤需在暗处进行:(3)PBS浸洗细胞3次,吸干,滴加DAPI 37℃避光孵育5 min进行细胞核染色;(4)PBS 清洗3次,吸干多余液体,自然干燥后,荧光显微镜下观察实验结果;(5)采用PhotoshopCS3软件将融合蛋白荧光图谱与对应的细胞核荧光图谱进行Merge,通过观察分析两者的荧光共定位情况,判断融合蛋白的亚细胞定位。
1.2.6 qRT-PCR检测BLM基因超表达后对Ran的影响 转染细胞48 h后,提取细胞总RNA,反转录后采用SYBR Green荧光染料法检测BLM和Ran基因的表达情况。荧光定量PCR反应体系:2×QuantiFast SYBR Green PCR Master Mix 5 μL、Sense Primer 0.5 μL、Anti-sense Primer 0.5 μL、cDNA 模 板 1 μL、ddH2O 3 μL、Total 10 μL,每个样品做 3 个重复 ;反应条件:95℃预变性2 min、95℃变性30 s、60℃退火30 s,反应进行40个循环后分析溶解曲线。随后,采用2-△△Ct方法对实时荧光定量PCR结果进行统计分析,2-△△Ct表示目的基因的相对表达量。最后采用SPSS Statistics 19.0 数据分析软件和GraphPad Prism 5 图形分析软件分别进行相关数据的统计分析和图表处理。
1.2.7 BLM蛋白在PC3细胞中表达量的检测 提取转染pEGFP-N3-BLM载体36 h后的PC3细胞总蛋白,以转染pEGFP-N3空载体的PC3细胞为对照,采用1.2.4中目标蛋白分离鉴定方法分离BLM蛋白,同样方法转膜封闭,随后以抗BLM蛋白小鼠单克隆抗体为一抗4℃孵育过夜,HRP山羊抗小鼠二抗孵育1 h,ECL显色,荧光化学发光成像系统分析BLM蛋白在PC3细胞中的表达情况。
2 结果
2.1 pMD-19-BLM克隆载体的鉴定
以B1、B2、B3三段序列的连接产物为模板,扩增BLM基因 CDS序列,胶回收后连接pMD19载体,转化JM109细胞。PCR检测和双酶切检测结果分别见图3。由图3-A可见,6个质粒在4 251 bp处均出现预期条带;由图3-B可见,KpnⅠ与XhoⅠ进行双酶切后,结果1-4泳道均在4 251 bp和2 692 bp左右出现两条亮带。经上海英骏生物技术公司对4个质粒进行测序鉴定,测序结果经MegAlign分析表明,3、4号质粒BLM基因序列发生了移码突变,2号质粒尽在aa192位处发生有义突变,由天冬酰胺(N)突变为天冬氨酸(D),其它均为无义突变,与GenBank中公布的BLM氨基酸序列(NM_000057)同源性最高。由于天冬酰胺属于极性中性氨基酸,天冬氨酸属于酸性氨基酸,两者所带R基团均具有一定的亲水性,因此该氨基酸的突变对BLM解旋酶蛋白功能的影响不大,所以保藏2号质粒对应菌株,作为后期构建原核和真核表达载体的中间载体。
2.2 重组原核和重组真核表达载体的鉴定
将连接好的重组原核和重组真核表达载体分别转化BL21和JM109细胞后,挑取阳性克隆进行培养,并提取质粒进行双酶切鉴定,结果见图4。由图4-A可见,仅6号泳道质粒在5 900 bp和4 251 bp出现目的条带,大小符合pET-32a-BLM酶切图谱,说明6号质粒酶切成功。由图4-B可见,1-5号泳道,均在4 700 bp和4251 bp出现两条目的条带,大小符合pEGFP-N3-BLM酶切图谱。对酶切成功的原核和真核表达质粒进行测序鉴定,并经MegAlign序列比对软件进行分析,结果显示重组原核和重组真核表达载体与前期构建成功的pMD19-BLM亚克隆载体序列同源性达100%,均可用于后期蛋白质的表达实验。

图3 转染JM109感受态细胞后PCR和双酶切检测pMD19-BLM

图4 pET32-BLM和pEGFP-BLM表达载体的双酶切检测
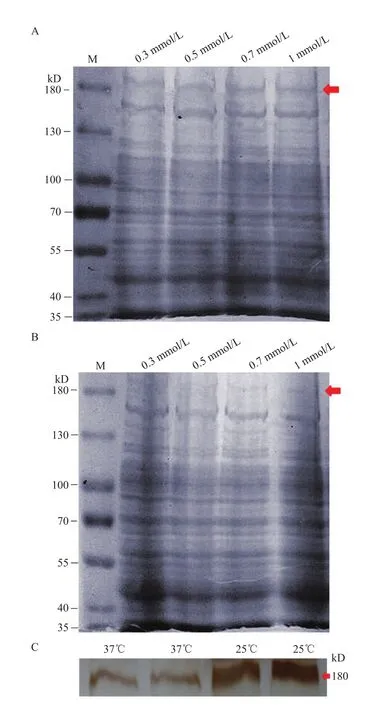
图5 BLM解旋酶在BL21中的表达
2.3 BLM解旋酶的在大肠杆菌中的表达结果
为研究BLM解旋酶在大肠杆菌中的最优表达条件,实验设置了5个IPTG浓度,两个培养温度,结果(图5)发现IPTG终浓度对BLM解旋酶的表达影响不大,而温度对BLM解旋酶的表达影响较大,且在25℃时表达量较高。Western-blot检测结果(图5-C)表明,构建的pET-32a-BLM原核表达载体能够在大肠杆菌中正确表达,且在25℃时表达量较高。
2.4 BLM基因的亚细胞定位
PSORT II Prediction分析软件对BLM解旋酶氨基酸分析(表 2)表明,BLM蛋白87.0%集中在细胞核中表达,4.3%在细胞质中表达,4.3%在细胞骨架中表达,4.3%在质膜中表达。经细胞转染,DAPI染核处理,BLM解旋酶的荧光共定位结果(图6)可见,GFP蛋白在整个中均有表达,而GFP-BLM融合蛋白主要集中在细胞核中表达,说明BLM主要在细胞核中表达,这与软件预测结果相同。
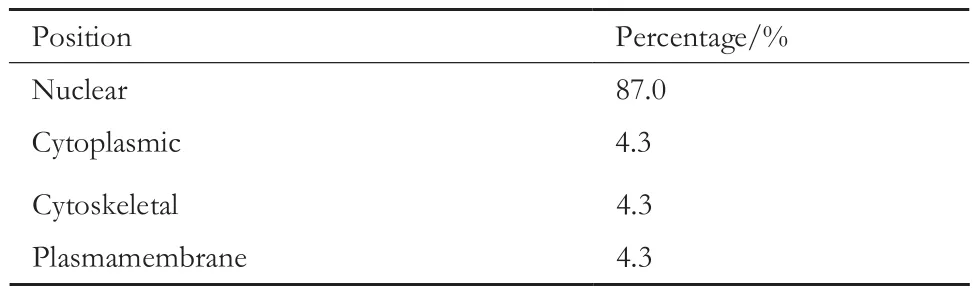
表2 PSORT II Prediction软件分析BLM蛋白的亚细胞定位
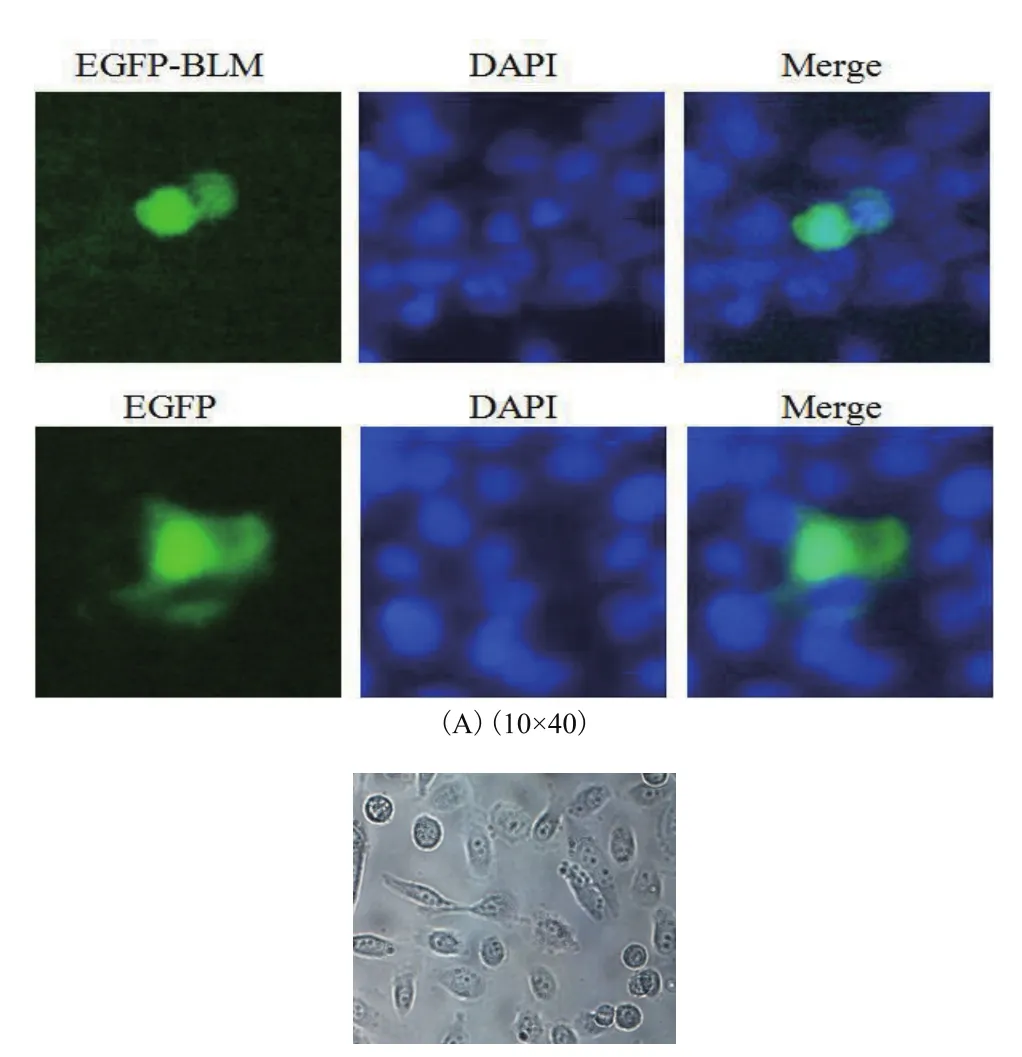
图6 BLM解旋酶在PC3细胞中的荧光定位图
2.5 Western bloting结果和荧光定量PCR结果
Western bloting结果(图7-A)表明,转染pEGFP-N3-BLM后,与对照组相比,BLM解旋酶在翻译水平获得了超表达。荧光定量PCR结果(图7-B)也表明,BLM基因在转录水平与对照组相比实现了超标达,且达到了极显著性差异(P<0.01);实验组与对照组相比,蛋白入核相关基因Ran表达量下降,与对照组相比也达到了极显著性差异(P<0.01)。

图7 BLM超表达后相关基因在转录水平和翻译水平的表达情况
3 讨论
在人类5种RECQ解旋酶中,BLM、WRN、RECQ4基因的突变分别会导致布鲁姆综合征(Bloom syndrome,BS)、 沃 纳 综 合 征(Werner syndrome,WS)和先天性血管萎缩皮肤异色综合征(Rothmund Thomson syndrome,RTS)3 种不同的疾病[2],其中BS患者最典型的特点是细胞中姐妹染色单体交换率(Sister chromatid exchanges,SECs)比正常人高出10倍。BLM基因编码区为4 251 bp,编码1 417个氨基酸残基,蛋白分子量约为159 kD,由于全蛋白分子量大,难以表达纯化,因此关于BLM全酶活性的研究一直受到限制。BLM639-1290区段含有Helicase、RecQ-Ct和HRDC结构域,Helicase的主要功能是负责DNA的结合、易位、解链和偶联ATP水解过程;RecQ-Ct作为RecQ解旋酶家族所特有的结构域,具有DNA结合和解链的活性,也与蛋白质折叠有关,但并不结合ATP;HRDC结构域能辅助性结合DNA进而调节其解链功能,也可增强和稳定BLM解旋酶与DNA的结合,但不具有催化活性[12]。因此,这3个结构域共同组成了BLM解旋酶的核心区,并具有全酶类似的活性,已成为研究者开展BLM全酶功能研究的首选对象。然而本课题组前期研究发现,在缺乏C端的BLM639-1290核心区,蛋白亚细胞定位主要集中在细胞质中,对BLM蛋白行使其主要功能造成较大影响。同时,最新研究也表明,BLM解旋酶N端关键氨基酸残基的磷酸化、泛素化和类泛素化修饰在修复损伤的DNA[13-15]、增强蛋白间的相互作用维持蛋白质的稳定性[16-17]、维持基因组(包括端粒和染色体)稳定性[18-19]、促进细胞凋亡[4]等方面具有重要作用。可见BLM639-1290核心区在开展体内功能验证实验时,不能准确反应BLM全蛋白的相关功能。
因此,本研究通过分段扩增的方法,旨在克隆BLM解旋酶全蛋白。结果成功克隆了人BLM解旋酶全CDS区,构建了人BLM基因的重组原核表达载体pET-32a-BLM和重组真核表达载体pEGFP-N3-BLM。原核表达结果发现,不同IPTG浓度对BLM蛋白的表达影响不大,低温有助于BLM蛋白表达量的提高,但在室温环境25℃、摇床转速180 r/min条件下,BLM蛋白主要以包涵体形式存在。研究表明,外源基因在大肠杆菌中的表达与多重因素有关,例如蛋白分子量的大小、培养温度、摇床转速、IPTG浓度以及培养液营养物质的丰富程度等,低温、低转速及适当降低培养基营养成分均有助于蛋白质的可溶性表达,因为在这些条件下,细菌生长缓慢,不易形成包涵体。因此,根据论文实验结果,后期应在同一IPTG浓度下,开展不同低温条件、不同低摇床转速、及不同营养成分培养基条件下的蛋白表达条件优化实验。
真核表达结果发现,PSORT II Prediction软件和亚细胞定位结果均表明BLM全蛋白的亚细胞定位主要集中在细胞核中,部分在细胞质中,其中细胞核中占87.0%,细胞质中占4.3%,细胞骨架中占4.3%,质膜中占4.3%;这与该蛋白在细胞质核糖体中合成,随后转运至细胞核中行使其主要生理功能相吻合。此外,为进一步证明BLM蛋白主要在细胞核中表达,本研究通过荧光定量PCR试验,检测入核关键蛋白Ran的表达情况。Ran 蛋白是一种 G 蛋白,与单分子鸟嘌呤核苷酸形成结合体(Ran-GTP)调节被转运物复合体的组装和解体,在细胞核中释放出携带NLS 的靶蛋白[20]。结果发现转染pEGFP-N3-BLM质粒48 h后,BLM基因实现了超表达,而Ran基因表达量却有所下降。分析原因,可能主要与检测时间点的选择有关。转染后48 h虽然BLM仍处于高表达状态,但细胞为维持自身正常生命代谢,可能会通过调节Ran蛋白的表达量来阻止BLM的持续入核,因此在48 h 检测Ran基因,其表达量在转录水平会明显降低。Western Blotting检测证实构建的真核表达载体和原核表达载体均能在PC3细胞和BL21细胞中表达出一个分子量约为179 kD 的蛋白质,与预测分子量大小基本一致,说明构建的两个表达载体正确无误。
4 结论
成功克隆了BLM基因CDS区,构建了pEGFPN3-BLM真核表达载体和pET-32a-BLM原核表达载体,并通过亚细胞定位实验证明了BLM蛋白主要在细胞核中表达,Western Blotting实验证实论文构建的原核表达载体和真核表达载体均能在相应的宿主细胞中正确表达。
