STXBP1基因突变致癫性脑病8例病例系列报告
2018-11-08杜晓楠吴冰冰李春培
路 通 杜晓楠 吴冰冰 周 浩 李春培 王 艺

1 方法
1.2 资料截取 使用ICD-10编码(G41.900)及关键词通过门诊信息系统查询病案资料,截取符合纳入标准病例的性别、年龄、起病年龄、发作类型、病程、出生史、生长发育史、家族史、就诊时体格检查、辅助检查、智力发育筛查(DST)结果、Gesell发育诊断量表评估结果、基因检测结果、诊断、治疗和随访情况。
2 结果
2.3 体格检查 8例均有不同程度的反应欠佳、眼示踪较差;例5、7表现为角弓反张;例3皮肤黝黑,阴囊着色较深;无特殊面容或小头畸形等。
表1 8例STXBP1基因突变致癫脑病患儿的临床资料
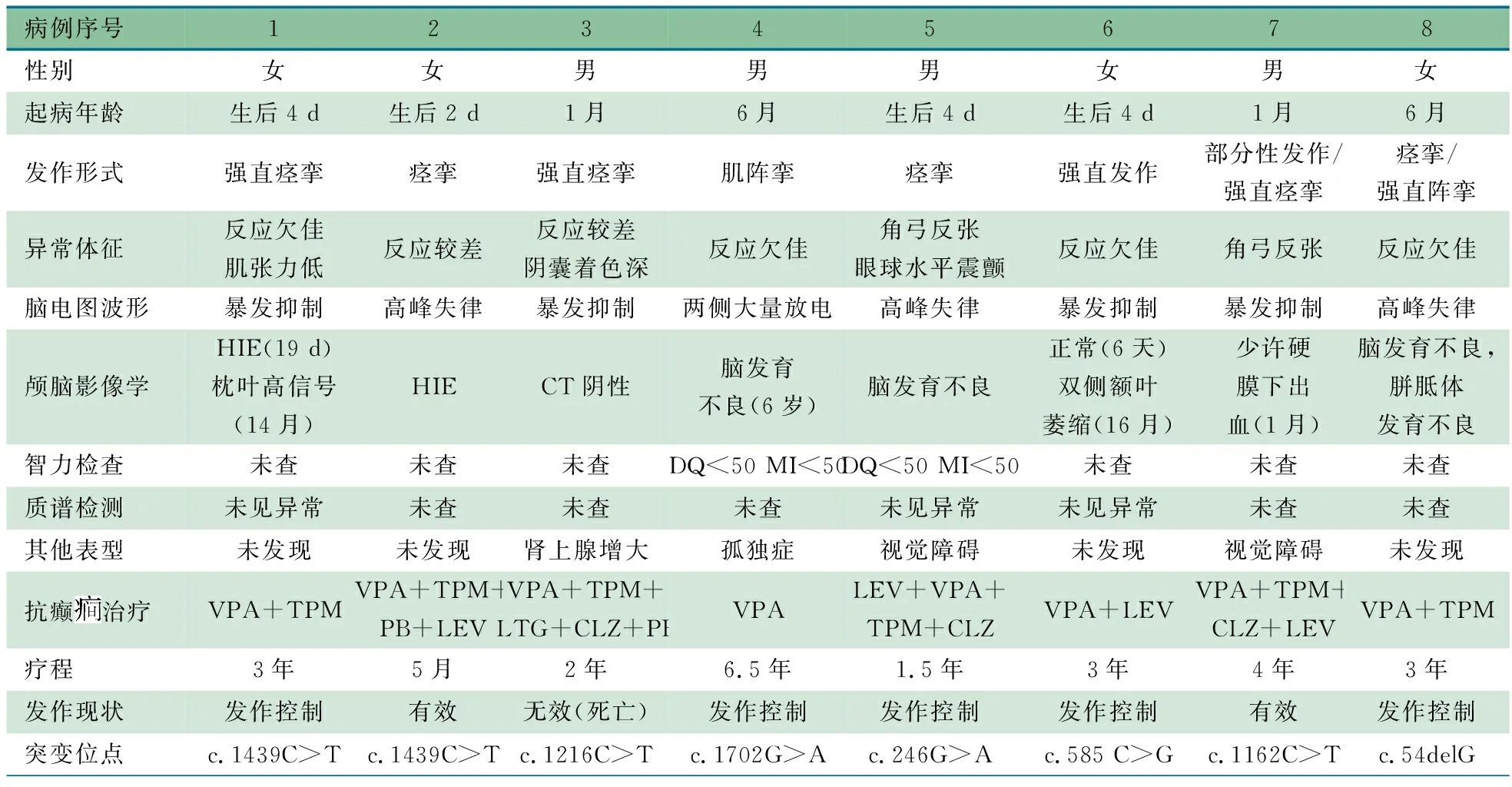
表1 8例STXBP1基因突变致癫脑病患儿的临床资料
病例序号12345678性别女女男男男女男女起病年龄生后4 d生后2 d1月6月生后4 d生后4 d1月6月发作形式强直痉挛痉挛强直痉挛肌阵挛痉挛强直发作部分性发作/强直痉挛痉挛/强直阵挛异常体征反应欠佳肌张力低反应较差反应较差阴囊着色深反应欠佳角弓反张眼球水平震颤反应欠佳角弓反张 反应欠佳脑电图波形暴发抑制高峰失律暴发抑制两侧大量放电高峰失律暴发抑制暴发抑制高峰失律颅脑影像学HIE(19 d)枕叶高信号(14月)HIECT阴性脑发育不良(6岁)脑发育不良正常(6天)双侧额叶萎缩(16月)少许硬膜下出血(1月)脑发育不良,胼胝体发育不良智力检查未查未查未查DQ<50 MI<50DQ<50 MI<50未查未查未查质谱检测未见异常未查未查未查未见异常未见异常未查未查其他表型未发现未发现肾上腺增大孤独症视觉障碍未发现视觉障碍未发现抗癫治疗VPA+TPMVPA+TPM+PB+LEVVPA+TPM+LTG+CLZ+PBVPALEV+VPA+TPM+CLZVPA+LEVVPA+TPM+CLZ+LEVVPA+TPM疗程3年5月2年6.5年1.5年3年4年3年发作现状发作控制有效无效(死亡)发作控制发作控制发作控制有效发作控制突变位点c.1439C>Tc.1439C>Tc.1216C>Tc.1702G>Ac.246G>Ac.585 C>Gc.1162C>Tc.54delG
注 HIE:缺氧缺血性脑损伤;VPA:丙戊酸钠; TPM:托吡酯;LEV:左乙拉西坦; CLZ:氯硝西泮;LTG:拉莫三嗪;PB:苯巴比妥
2.4 辅助检查 ①颅脑影像学检查:7例行MR,例4、5、8为脑发育不良,例1为新生儿缺血缺氧性脑病、枕叶高信号,例6为枕叶萎缩与侧脑室增宽,例7为少许硬膜下出血。例3行颅脑CT检查未见异常。②脑电图:7例行睡眠脑电图、例4行视频脑电图检查,例1、3、6、7为暴发抑制波形,例2、5、8为高峰失律波形,例4为两侧较多痫样放电,例3、5、6的脑电图表现分别见图1A、B、C。③智力:2例行DST, DQ(发育商)均<50,MI(智商指数)均<50。④其他:例3行喉镜检查显示合并先天性喉软骨发育不良,腹部B超示肾上腺皮质均质性增大(图2)。例4患儿孤独症量表提示孤独症谱系障碍。例5、7行视觉诱发电位检测,提示双眼波形分化差,听觉诱发电位检测无明显异常。例1、5、6行质谱检测未见异常。例1、6行心超检查,未见异常。

图1患儿的脑电图波形

图2例3肾上腺B超图像
注 A:右侧肾上腺均质增大, B:左侧肾上腺均质增大,如图中红色箭头所示
2.5 基因检测 表2显示,8例共检测到7个STXBP1基因突变位点,其中错义突变4个,无义突变2个,移码突变1个。错义突变经过SIFT、Polyphen-2和Mutation Taster预测均为有害突变。4个突变位点c.1216C>T、c.1702G>A、c.246G>A、c.54delG尚未被HGMD收录。家系验证结果显示,8例均为新发突变。

表2 8例患儿的STXBP1基因突变信息
注 1)该位点为识别剪切位点的错义突变,2)移码突变造成后续氨基酸序列改变,3)不适用。SIFT评分0~1,<0.05分推测有害,≥0.05分推测为可容忍;Polyphen2评价结果主要分“良性”和“可能有害”;Mutation taster结果中P值代表预测的可信度,越接近1,准确度越高
3 讨论
STXBP1基因位于9q34.11,编码STXBP1(MUNC18-1)蛋白,其最长的转录本有20个外显子[7]。STXBP1基因在哺乳动物神经系统广泛表达,在进化上高度保守。STXBP蛋白与Syntaxin-1蛋白协同参与可溶性NSF附着蛋白受体(SNARE)核心复合体的装配,通过与突触融合蛋白(SYNTAXIN)相互作用,参与SNARE核心复合体的形成以及突触小泡与突触前膜的融合,影响神经递质的释放[8]。
文献报道中STXBP1基因突变其他的神经系统异常主要包括肌张力低下,共济失调,震颤,肌张力障碍等[14]。合并精神行为异常的主要表型为孤独症谱系障碍(ASD)[15, 16],其比例约为20%。本文例4合并孤独症。例3阴囊着色深,皮肤偏黑,腹部B超提示肾上腺均质性增大。已有文献报道2例STXBP1基因拷贝数缺失的患儿有肾脏异常,1例为右肾发育不良[17],1例为肾盂扩张[18],提示该异常可能为STXBP1基因突变新的表型。
既往文献中,携带STXBP1基因突变的患儿中50%以上有异常的脑电图表现,最常见的表现为暴发抑制和高峰失律。本文8例中, 4例表现为暴发抑制; 3例表现为高峰失律;1例存在局灶及多灶性放电,棘慢波,多棘波,阵发异常电活动,脑电背景变慢。
本文8例患儿中共检测到STXBP1基因7个不同的突变位点, 其中4个突变位点为首次报告,3例错义突变c.1216C>T(p.R406C)、c.246G>A(p.K82K)和c.1702G>A(p.G568S),经过SIFT、Polyphen-2和Mutation Taster软件预测均为有害变异,另1例移码突变c.54delG会造成后续氨基酸序列的改变而影响蛋白的功能,结合患儿的临床表现与辅助检查结果,推测这4个位点均为致病突变。
