慢性进行性眼外肌麻痹5型合并假肥大型肌营养不良征象患者临床与遗传学特点
2018-10-30宋莉谢友娜黄芷琳何柏萱裴中姚晓黎
宋莉 谢友娜 黄芷琳 何柏萱 裴中 姚晓黎
常染色体显性遗传慢性进行性眼外肌麻痹(autosomal dominant chronic progressive external ophthalmoplegia,ad CPEO)5 型(PEOA5)作为核基因遗传性线粒体病的罕见类型,是一种和 mtDNA多重缺失有关的线粒体脑肌病。以进行性眼球运动受限和上眼睑下垂为主要表现,部分伴有咽部和四肢近端的肌肉麻痹[1]。假肥大型肌营养不良(duchenne/becker muscular dystrophy,DMD/BMD)以缓慢进展的四肢近端骨骼肌对称性肌萎缩无力、小腿腓肠肌假性肥大为特征,可伴不同程度的认知障碍和心肌、呼吸肌损害[2]。目前线粒体性肌病合并肌营养不良病变的报道非常罕见[3],相关指标及遗传学研究亦非常缺乏,本研究发现一例罕见的ad CEPO合并BMD征象患者,并进行了初步的遗传学研究。
1 病例资料
1.1 先证者资料先证者,男,13岁,双睑下垂13年,发现肌酶升高10年,四肢无力2年。患者35周剖宫产,出生时体重2.1 kg,发现患儿双眼睑下垂,但哭声响亮,进食、活动无异常,3+个月抬头,8个月会坐、会笑,10个月站、说话、认人,15个月独立走路,此后运动功能与同龄儿相仿。3岁时体检发现转氨酶增高,可跑跳,当地医院查体见腓肠肌稍饱满,膝反射稍弱,从仰卧位站立时需双手撑床面,未行特殊治疗。7岁上小学,体育课上跑步比同学慢。近2年出现进行性四肢无力,双上肢不能提重物,上楼困难,下蹲起立困难,跌倒频繁,症状逐渐进展,从行走1 h需休息到目前行走500 m需要休息。近2年出现持续性复视。所有症状无晨轻暮重表现,无吞咽困难、饮水呛咳和构音障碍,无呼吸困难,无肌痛,无视力下降,无抽搐、肢体麻木。神经系统查体:高级神经活动正常,计算力、定向力、认知正常,神清,言语流利,双眼睑下垂,左眼8 mm,右眼6 mm,双眼向下活动受限,双侧鼻唇沟对称,伸舌居中,巨舌,无舌肌纤颤,双下肢近端肌肉萎缩,腓肠肌肥大,四肢肌张力正常,双上肢肌力4+级,双下肢近端肌力3级、远端肌力4级,四肢腱反射减低(+),双侧痛觉对称存在,双下肢病理征未引出。经醋酸泼尼松片15 mg、甲钴胺片、艾地苯醌等药物治疗1年,症状无好转。辅助检查:2006 年肌酶:CK4332 U/L,2007 年 CK 12196 U/L,2012年 CK 29485 U/L。2006年新斯的明试验(-);肌电图:肌源性损害活动期可能性大;肌活检(右腓肠肌):送检肌肉组织正常结构存在,肌束中可见少量呈大圆形透明变性的肌纤维,灶性炎症细胞浸润及肌纤维再生,肌束间纤维结缔组织和脂肪组织轻度增生,PAS染色未见异常物质贮积,Gomori染色未见破碎红纤维。2017年血乳酸 2.5 mmol/L(正常 0.7~2.0 mmol/L);心脏彩超:心脏结构及瓣膜活动未见明显异常。
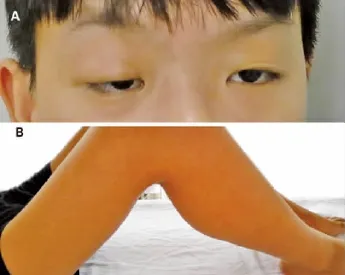
图1 A双眼睑下垂 B双下肢近端肌肉萎缩,腓肠肌肥大
1.2 先证者父母、弟弟资料父母亲均无四肢无力,无眼睑下垂,无复视,无腓肠肌肥大,父亲“乙肝小三阳”,父母非近亲结婚。弟弟半岁,无眼睑下垂、无四肢无力、无吞咽困难,曾行产前检查未发现相关致病基因。
1.3 基因检测运用二代测序(NGS)的方法对先证者的外周血样本进行骨骼肌疾病相关基因外显子捕获检测,发现其样本中RRM 2B基因外显子1上存在c.132G>A点突变(p.W44X)杂合突变及DMD基因外显子6上存在c.431T>A点突变(p.V144 D)半合子变异。然后对其父母c.132G>A位点进行验证发现,母亲携带变异,父亲未携带变异。C.431T>A位点验证,父亲母亲均未携带有变异。其中RRM2Bc.132G>A为无义突变,临床意义未明,DMDc.431T>A为错义突变,为疑似致病性变异,两突变均不属于多态性位点,尚没有文献报告,为新发现的变异。RRM2B、DMD基因分析结果如下:
1.4 家系图结合基因分析,得出家系图,该家系RRM 2B呈常染色体显性遗传,DMD为散发突变(图 3)。
2 讨论
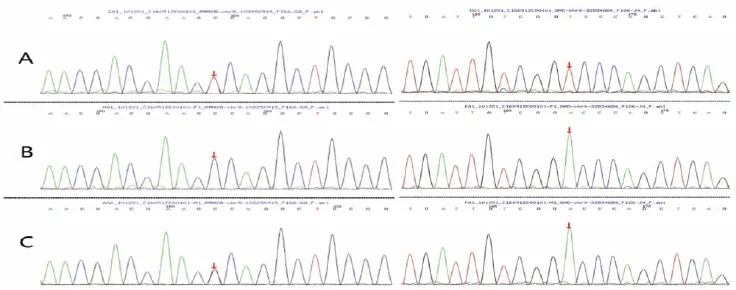
图2 基因分析结果:A为先证者,左RRM 2B基因c.132G>A杂合变异,右DMD基因c.431T>A半合子变异;B为先证者父亲,基因分析结果无变异;C为先证者母亲,左RRM 2B基因c.132G>A杂合变异,右DMD基因c.431T>A无变异
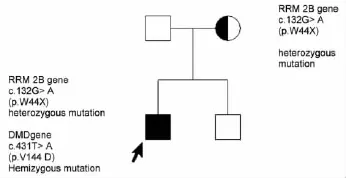
图3 家系的系谱图
CPEO是一种和mtDNA多重缺失有关的线粒体脑肌病。目前研究表明CPEO多数由线粒体DNA单一大片段缺失或A3243G点突变造成,少数与核DNA突变有关[4]。成年线粒体病约一半由于核基因突变引起。相关研究数据显示核苷酸还原酶RRM2B是继POLG(27%)和TWINKLE(14%)之后第三个与成人线粒体DNA缺失有关的最常见原因(13%)[5]。RRM2B基因定位于8号染色体长臂(8 q23.3),全长 4955 kb。 Bourdon A.等[6]第一次报告纯合子RRM2B的丢失突变是婴幼儿导致严重mtDNA消耗综合征(mitochondrial DNA depletion syndrome,MDS)的一个原因。 早发性的MDS多呈现为RRM2B隐性遗传突变,而RRM 2B显性遗传基因突变具有临床异质性,常见于致命的小儿神经肌肉伴肾小管功能不全综合征[7]、晚发型ad CPEO等。一般来说,ad CPEO的肌病表型相对良性,发病年龄≥20岁,童年期症状相对稳定,成人ad CPEO可出现上睑下垂和眼肌瘫痪,伴随着轻微的肌肉疲劳等症状,但其血清CK正常或轻度升高[8-9]。假肥大型肌营养不良(duchenne/becker muscular dystrophy,DMD/BMD)是儿童遗传性肌病的一个主要类型,临床上大多数病例有明确的家族史,约1/3为散发性病例。研究证实,DMD/BMD均是由于抗肌萎缩蛋白基因 (即DMD基因)突变所致[10]。DMD基因是人类最大的基因之一,定位于X染色体短臂 (Xp 21.1-3),全长2500 kb,由79个外显子和78个内含子构成。DMD基因编码3685个氨基酸,组成427 kD细胞骨架蛋白-Dystrophin蛋白。Dystrophin蛋白缺乏可造成肌细胞膜不稳定,肌肉收缩时出现各种机械因素的损伤,导致肌纤维坏死和功能缺失而发病[11]。BMD被认为是DMD的一种不典型类型,症状与之相似,但发病较晚,症状较轻,心脏少受累,少数可接近正常生命年限。
本研究患者3岁仰卧位站立需双手撑床面、腓肠肌轻度肥大,11岁之后开始出现双下肢无力,下蹲困难,跌倒频繁,13岁双下肢肌力3~4级,近端无力萎缩明显,双侧腓肠肌轻度肥大,腱反射减弱,CK高达29485U/L,肌电图提示肌源性损害,基因检测为RRM2B基因外显子1上存在c.132G>A无义突变,氨基酸p.W44X提前出现终止密码子致病可能性大,家系系谱图分析呈杂合突变发病,推测遗传方式呈现常染色体显性遗传,RRM 2B基因的有害致病性突变已被报道可导致PEOA5,因此可以诊断是PEOA 5型。另一方面,患者基因检测DMD基因错义突变,该位点尚无文献报道致病,疑似致病性突变,可能导致氨基酸p.V144D功能改变,符合可能的DMD/BMD的诊断。
一般来说,CPEO血清CK仅正常或轻度升高,而DMD眼球运动、发音、吞咽不受累。本研究患儿临床表现有ad CPEO、BMD部分特征,难以用单独疾病解释所有症状。KIYOMOTO[3]等总结了86例肌活检证实的CPEO患者,其中3例发现线粒体肌病合并肌营养不良的特性,表现亦为骨骼肌近端无力,并CK显著升高。原因可能与核基因修饰符加重疾病状态相关,但其具体机制仍有待阐明。本研究的基因检查在同一个病人身上同时发现两种疾病相关基因的致病性突变或可疑致病性突变位点,目前尚无相关文献报道,属新发现的致病性突变位点。两种基因编码的蛋白对其生物学功能的影响是否有协同或修饰作用尚有待进一步研究。由于患者3岁时肌活检未发现破碎红纤维及dystrophin功能缺损,且近2年出现肌无力后无法进一步检查,故诊断缺乏肌肉活检的支持。但遗传学及基因测序检查帮助明确诊断,发挥了极其重要的作用。
综上所述,本家系患者可能的发病机制:① RRM 2B基因8号染色体1号外显子上存在c.132G>A点突变,编码区第132号核苷酸由鸟嘌呤异变为腺嘌呤,导致该基因编码的第44号密码子由色氨酸变为终止密码子,为无义突变,提前终止多肽链合成,造成蛋白功能缺失,影响线粒体内的脱氧核苷酸“池”的平衡,导致mtDNA合成的原料供应障碍,从而影响mtDNA复制,进而降低线粒体的能量代谢功能[12]。同时,核基因的修饰作用、mtNDA多重缺失可能导致肌营养不良征象出现。②DMD基因X染色体6号外显子c.431T>A点突变,编码区第431号核苷酸由胸腺嘧啶异变为腺嘌呤,导致该基因编码的第144号密码子由缬氨酸变异为天冬氨酸,为错义突变,可能造成蛋白功能改变,造成肌细胞膜不稳定,导致肌纤维坏死和功能缺失。③母亲RRM 2B基因c.132G>A杂合变异,可推测该突变可能来自母亲,患者杂合突变发病,推测遗传方式呈常染色体显性遗传,母亲无临床症状,推测可能与基因不全外显有关。DMD基因c.431T>A半合子变异,父母均未携带有该变异,推测为自发突变。近年来,CPEO及DMD相关基因突变多有报道[13-15],上述2个基因的突变位点分属无义和错义突变,尚无相关文献报道,是新发现的突变,根据基因有突变等级的判定标准[16],应该属致病性突变和可疑致病性突变。
3 结论
线粒体性肌病与肌营养不良特征性病变共存的报道非常罕见,两种疾病致病基因是否有协同作用或核基因的修饰作用、mtNDA多重缺失可能导致线粒体肌病中肌营养不良征象,机制尚需进一步探讨阐明。本研究发现的ad CPEO及DMD基因突变属于尚未报道的可能致病性突变位点,说明基因分析是确诊CPEO、BMD等疾病的可靠方法。对临床上进展性眼睑下垂合并不典型肌无力的患者,在进行肌肉活检的基础上开展神经肌肉病的相关基因分析,可能有助于早期诊断不同肌病类型及其具体分型。
