迪拉马尼的合成研究
2018-10-15郑雪敏宋艳玲
郑雪敏, 卢 鑫, 宋艳玲
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
迪拉马尼(Delamanid,1,又名地依麦迪),化学名为(2R)-2,3-二氢-2-甲基-6-硝基-2-[[4-[4-[4-(三氟甲氧基)苯氧基]-1-哌啶基]苯氧基]甲基]咪唑并[2,1-b]噁唑,分子式为C25H25F3N4O6,相对分子质量为534.48.其由日本Otsuka公司研发,通过干扰结核分枝杆菌(MTB)细胞壁的新陈代谢达到对多重耐药肺结核病(MDR-TB)的治疗[1].迪拉马尼是近40年来第2个获批治疗MDR-TB的新型药物,于2014年5月经欧洲药品管理局(EMA)批准上市,商品名为DELTYBA[2].由于其疗效显著,不良反应发生率低,应用前景广阔,因此,本文设计和优化了一种迪拉马尼合成路线.该路线操作简便,生产周期短,产品纯度和收率均较高,成本较低,易于放大生产,能在一定程度上降低迪拉马尼原料药的生产成本,从而对其成品药价格的进一步降低起到积极促进作用,有利于减轻患者的经济负担.
文献[3-7]关于迪拉马尼的合成报道均是以4-[4-(4-三氟甲氧基苯氧基)哌啶-1-基]苯酚(3)和(R)-4-硝基苯磺酸-2-甲基缩水甘油酯(6)为关键中间体,合成路线见图1.路线一:化合物3和6通过亲核取代反应得到(R)-1-[4-(2,3-环氧-2-甲基丙氧基)-基]-4-(4-三氟甲氧基苯氧基)哌啶(7),7 与2-溴-4-硝基-1H-咪唑经亲核取代及环合反应一勺烩制得1,收率为17.7 %(以β-甲基烯丙醇为起始原料计算);张志国[7]等对该路线进行优化,收率提高至30 %(摩尔收率).路线二:6 与2-溴-4-硝基咪唑反应得(R)-2-溴-1-(2-甲基-2,3-环氧丙基)-4-硝基-1H-咪唑(8),8 与3 经亲核取代反应及环合反应一勺烩制得1,收率为17.7 %(以β-甲基烯丙醇为起始原料计算),其中,3 是通过4-[4-(三氟甲氧基)苯氧基]哌啶(2)与1,4-环己二酮芳构化制得.
文献报道的这两种合成路线反应总收率较低,不利于工业放大.因此,本研究对迪拉马尼的新合成路线进行研究,参考文献资料[3-6],设计了以β-甲基烯丙醇(4)和4-[4-(三氟甲氧基)苯氧基]哌啶(2)为起始原料,经环化、置换、取代、磺化、缩合5步反应得到目标化合物.该路线简单有效,反应条件温和,反应总收率43.7 %(摩尔收率).具体合成路线见图2.
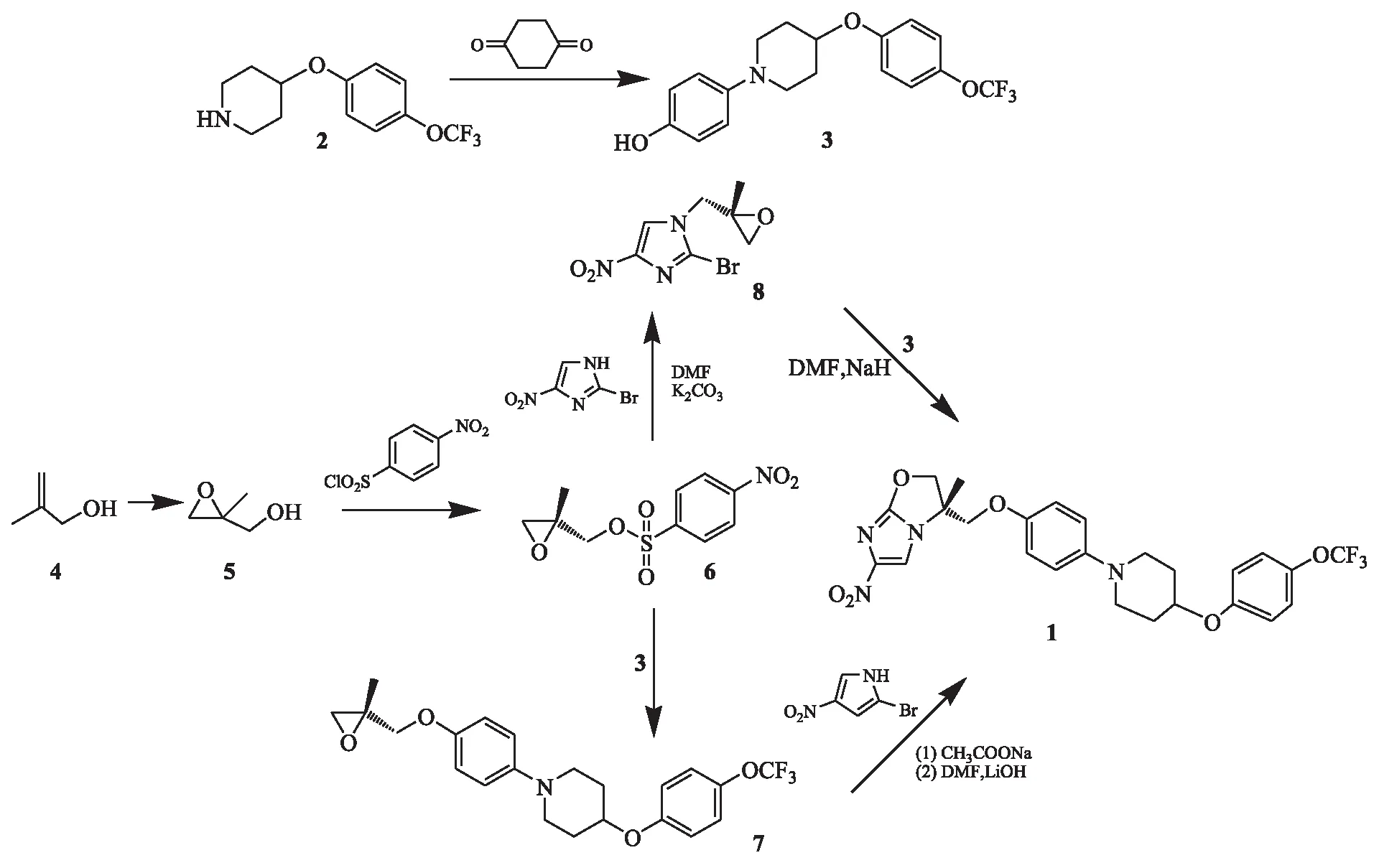
图1 迪拉马尼的合成路线
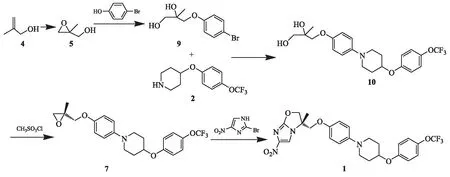
图2 迪拉马尼的合成新路线
1 实验部分
1.1 主要试剂和仪器
WRX-4型显微熔点仪,宁波科诚仪器有限公司,温度计未经校正;Mercury-300(300 MHz)核磁共振仪,美国Varian公司,TMS为内标;LCQ Advantage MAX 10离子肼质谱仪,美国菲尼根公司;高相液相色谱仪,日本岛津公司;实验所用试剂均为市售分析纯.
1.2 实验方法
1.2.1 S-2-甲基缩水甘油(5)的制备
在500 mL无水甲苯中依次投入β-甲基烯丙醇(60 g,0.75 mol),D-酒石酸二异丙酯(12.7 g,54 mmol),4A分子筛30 g,混合物搅拌5 min后,加入四异丙醇钛(13.5 g,47.8 mmol),搅拌1 h,降温至-15 ℃,滴加80 %(质量分数)的氢过氧化枯烯(40 g,263.2 mmol),约2 h滴加完毕,于-10 ℃下搅拌5 h;升至室温,滴加三氯氧磷(95.7 g,625.1 mmol),约1 h滴完,10~20 ℃反应4 h,静置过夜;加入硅藻土12 g后过滤,得S-2-甲基缩水甘油醇的甲苯溶液,直接投于下步反应.
1.2.2 R-3-(4-溴苯氧基)-2-甲基-1,2-丙二醇(9)的制备
向上述溶液中加入4-溴苯酚(108 g,0.6 mol)和100 mL质量分数为25 %的氢氧化钠溶液,在40 ℃搅拌反应9 h,冷却后静置过夜;加入活性炭9 g,搅拌1 h,过滤,分离苯层,饱和盐水洗涤,水洗、减压浓缩除去甲苯;降至室温,加入160 mL正己烷,搅拌析晶,过滤,100 mL冷的正己烷洗涤,得白色晶体,50 ℃干燥,得产物104.9 g.以β-甲基烯丙醇计算,收率为82.7 %(摩尔收率),mp 86~88 ℃.MS(m/z):262[M+H]+.1H-NMR(300 MHz,CDCl3):7.12(d,J=8.0 Hz,2H),6.89(d,J=8.0 Hz,2H),4.24~4.36(m,1H),3.11~3.17(m,2H),2.70~2.76(m,2H),1.94~2.06(m,2H),1.58~1.73(m,2H),1.54(brs,1H).
1.2.3 R-2-甲基-3-(4-{4-[4-(三氟甲氧基)苯氧基]哌啶-1-基}苯氧基)-1,2-丙二醇(10)的制备
将化合物(9)(60 g,243.6 mmol),4-[4-(三氟甲氧基)苯氧基]哌啶(60 g,217.2 mmol),三(二亚苄基丙酮)二钯(520 mg,0.52 mmol),膦配体(610 mg,0.47 mmol),叔丁醇钠(26.2 g,287.9 mmol)溶于180 mL甲苯中,氮气保护下,70 ℃反应10 h,冷却后加入氯化铵溶液,甲苯萃取、干燥、减压浓缩得化合物10,收率86 %(摩尔收率),mp 87~89 ℃,纯度98.3 %.[HPLC归一化法:色谱柱InertSustain-C18柱(4.6 mm×250 mm,5 μm);流动相:V(乙腈)∶V(水)=1∶9;10 mmol/L磷酸二氢钾为缓冲液,加磷酸调至pH 3.0;检测波长223 nm;柱温30 ℃;流速1.0 mL/min].MS(m/z):406[M+H]+.1H-NMR(300 MHz,CDCl3):1.28(s,3H),1.88~2.03(m,2H),2.03~2.19(m,2H),2.22(brs,1H),2.75(brs,1H),2.92~3.05(m,2H),3.30~3.45(m,2H),3.57(d,J=11.2 Hz,1H),3.73(d,J=11.2 Hz,1H),3.86(d,J= 9.0 Hz,1H),3.93(d,J=9.0 Hz,1H),4.36~4.48(m,1H),6.78~6.98(m,6H),7.13(d,J=9.3 Hz,2H).
1.2.4 R-1-[-4-[2,3-环氧-2-甲基丙氧基]苯基]-4-[4-(三氟甲氧基)苯氧基]哌啶(7)的制备
将化合物(10)(65 g,160 mmol)溶于300 mL乙酸乙酯,加入三乙胺(16.8 g,165 mmol),冰浴冷却下滴加甲基磺酰氯(19.2 g,165 mmol),反应完毕后,乙酸乙酯萃取、干燥、减压浓缩得(R)-2-羟基-2-甲基-3-(4-{4-[4-(三氟甲氧基)苯氧基]哌啶-1-基}苯氧基)丙基甲磺酸酯.将上述产物中加入600 mL甲醇、53.7 g碳酸钾,室温搅拌5 h,减压除去甲醇,加入甲苯和水搅拌,取甲苯层,水洗、干燥、减压浓缩,加入300 mL异丙醇,降温搅拌析晶,过滤、干燥,得化合物(7)62.7 g,收率92.3 %(摩尔收率),mp 82~84 ℃(文献[7]:84~85 ℃),纯度98.3 %.[HPLC 归一化法:色谱柱 InertSustain-C18柱(4.6 mm×250 mm,5 μm),流动相:V(乙腈)∶V(水)=1∶9;10 mmol/L磷酸二氢钾为缓冲液,加磷酸调至pH 3.0;检测波长223 nm;柱温30 ℃;流速1.0 mL/min].ee值98.6 %.[HPLC归一化法:色谱柱 Astec CHIROBIOTICTMTAM柱(4.6 mm×250 mm,5 μm);流动相∶V(正己烷)∶V(乙醇)∶V(二乙胺)=95∶5∶0.1;检测波长223 nm;柱温30 ℃;流速0.8 mL/min].MS(m/z):424[M+H]+.1H-NMR(300 MHz,CDCl3):7.12(d,J=8.4 Hz,2H),6.80~6.96(m,6H),4.39~4.48(m,1H),3.99(dd,J=25.8,10.5 Hz,2H),3.26~3.41(m,2H),2.97~3.01(m,2H),2.83(d,J=4.6 Hz,1H),2.75(d,J=4.9 Hz,1H),2.01~2.17(m,2H),1.82~2.07(m,2H),1.46(s,3H).
1.2.5 R-2-甲基-6-硝基-2-{4-[4-(4-三氟甲基苯氧基)哌啶-1-基]苯氧甲基}-2,3-二氢咪唑并(2,1-b)噁唑(1)的制备
将化合物(7)(10 g,23.6 mmol),2-溴-4-硝基咪唑(4.6 g,24.0 mmol),乙酸钠(0.4 g,32.4 mmol),10 mL乙酸叔丁酯在100 ℃下搅拌3.5 h,加入70 mL甲醇,滴加10 mL氢氧化钠溶液(质量分数为25 %),0 ℃下搅拌1.5 h,室温下搅拌4 h,加入15 mL水,5 mL乙酸乙酯,45~55 ℃搅拌1 h,冷却至室温,过滤产物,30 mL甲醇洗涤,40 mL水洗涤,100 mL异丙醇重结晶,降温至室温,过滤,甲醇洗涤,减压干燥得产物 9.3 g,收率73 %(摩尔收率),mp 195~196 ℃(文献[7] mp 195~196 ℃);纯度99.9 %.[HPLC归一化法:色谱柱 InertSustain-C18柱(4.6 mm×250 mm,5 μm),流动相:V(乙腈)∶V(水)=2∶8;10 mmol/L磷酸二氢钾为缓冲液,加磷酸调至pH 4.0;检测波长245 nm;柱温30 ℃;流速1.0 mL/min].ee值>99 % .[HPLC归一化法:色谱柱 Astec-CHIROBIOTICTMTAM柱(4.6 mm×250 mm,5 μm);流动相:V(正己烷)∶V(乙醇)∶V(二乙胺)=60∶40∶0.1,检测波长245 nm;柱温 30 ℃;流速:1.0 mL/min];MS(m/z):535[M+H]+.1H-NMR(300 MHz,CDCl3)δ:1.77(3H,s),1.8~2.2(4H,m),3.2~3.4(2H,m),3.9~4.1(2H,m),4.02(1H,d,J=10 Hz),4.04(1H,d,J=10 Hz),4.18(1H,d,J=10 Hz),4.4~4.5(1H,m),4.50(1H,d,J=10 Hz),6.78(2H,d,J=9 Hz),6.8~7.0(4H,m),7.14(2H,d,J=9 Hz),7.56(1H,s).
2 结果与讨论
2.1 结 果
以β-甲基烯丙醇(4)为起始原料,经环氧化反应和取代反应得到R-3-(4-溴苯氧基)-2-甲基-1,2-丙二醇(9),化合物 9 通过和 4-[4-(三氟甲氧基)苯氧基]哌啶(2)发生取代、磺化和缩合反应制得迪拉马尼(1).反应路线共5步,总产率约43.7 %(以4计算,摩尔收率).目标化合物结构经1H-NMR和ESI-MS确证,HPLC检测纯度﹥98 %,ee值﹥99 %.
2.2 讨 论
采用不对称合成法制备手性药物迪拉马尼,合成过程中S-2-甲基缩水甘油的合成尤为重要.以β-甲基烯丙醇为起始原料,四异丙醇钛作为手性催化剂,D-酒石酸二异丙酯作为手性源,氢过氧化枯烯作为氧化剂,通过Sharess不对称合成反应得到S-2-甲基缩水甘油.在反应体系中加入4A分子筛,可以对反应的产率和选择性有着积极的作用.反应溶剂的极性对反应的产率和选择性也具有一定影响,在此步反应中采用甲苯作为溶剂,得到较高ee值的产物.
3 结 论
迪拉马尼作为一种新型的治疗肺结核的药物,尤其适用于多耐药菌的患者.国内仿制药正处于开发中,此文对其合成路线进行研究,成功制备出了目标化合物,而且各项指标均与文献报道值相符.
已有的关于迪拉马尼制备的文献[3-7]报道中,均是以4-[4-(4-三氟甲氧基苯氧基)哌啶-1-基]苯酚(3)为重要中间体,化合物(3)是通过4-[4-(三氟甲氧基)苯氧基]哌啶(2)与1,4-环己二酮芳构化制得,反应收率约为50 %(摩尔收率).本研究设计的合成路线直接以化合物(2)作为起始原料,减少了反应步骤,反应总收率提高至43 %(摩尔收率)(文献[3-7]报道的收率为17 %~30 %(摩尔收率)),降低了生产成本,且反应操作简单,适合工业化生产.
