腹膜后Castleman病13例报告并文献复习
2018-10-10徐维锋周敬敏纪志刚
东 洁 徐维锋 周敬敏 谢 燚 纪志刚
(中国医学科学院北京协和医院泌尿外科,北京 100730)
Castleman病(Castleman’s disease,CD)是由美国病理学家Benjamin Castleman等在1956年首次报道的一种淋巴增生性疾病[1],也称巨大淋巴结病或血管滤泡性淋巴结增生症。国内外相关文献以个案报告为主。该病可发生于全身任何淋巴结区域,腹膜后Castleman病极为罕见,且缺乏特异性表现,临床上容易误诊、漏诊。现将我院2012年1月~2017年11月近6年收治的13例腹膜后Castleman病的临床资料进行回顾性分析,并复习相关文献,总结腹膜后Castleman病的临床特点及诊疗方法。
1 临床资料与方法
1.1 一般资料
检索2012年1月1日~2017年11月20日我院腹膜后肿物资料,经手术切除或组织活检石蜡病理确认Castleman病13例,男6例,女7例。年龄23~57岁,平均42岁。7例因体检超声偶然发现腹膜后肿物,3例因腰腹部隐痛不适就诊,3例因口腔、外阴溃疡及周身皮疹就诊。从出现症状或发现肿物到诊断Castleman病,确诊时间1~36个月,平均9个月。1例高血压病史13年(例5),1例乙肝病史20年(例12)。见表1。
7例术前行超声检查,肿瘤呈低回声实性占位,见表2。均行腹盆腔CT检查,肿瘤均呈软组织密度,5例肿瘤内有坏死或钙化,边界清,增强扫描肿瘤明显或中等强化(图1)。8例肿瘤周边伴多发淋巴结肿大,见表2。腹膜后病变部位多变,单中心12例,多中心1例(例12)。肿瘤最大径3.0~11.8 cm,平均6.0 cm。
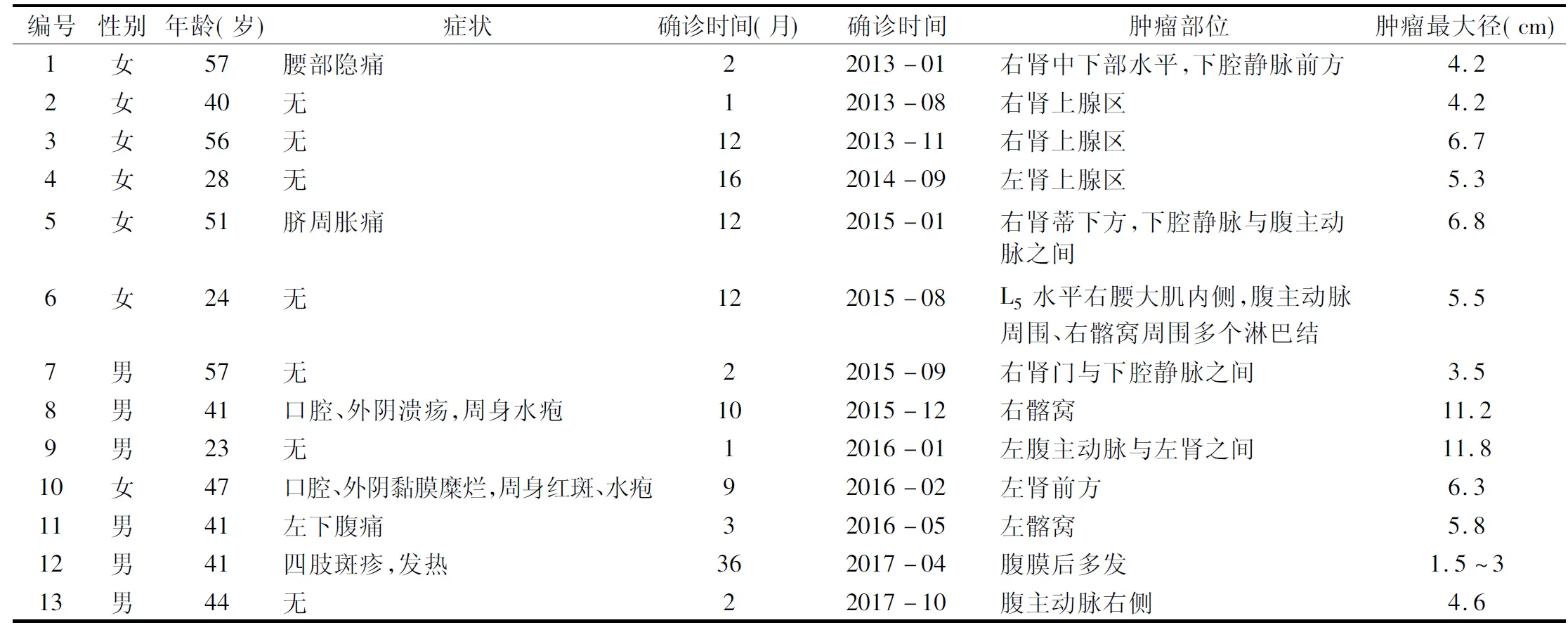
表1 13例一般资料
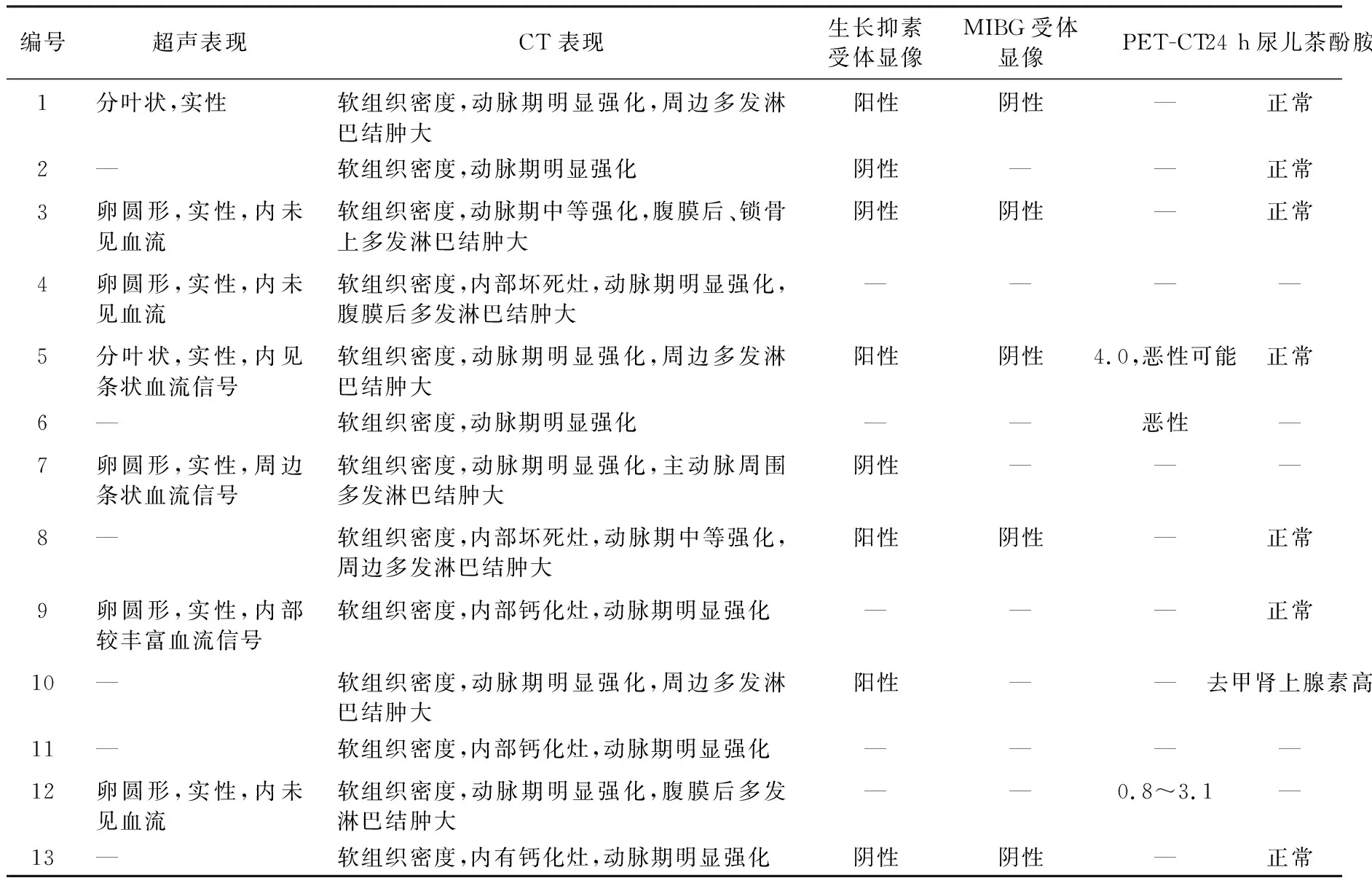
表2 13例影像学检查、核医学检查及化验检查结果
8例行生长抑素受体显像,其中阳性4例,阴性4例。5例行间碘苄胍(MIBG)肾上腺髓质受体显像,均为阴性。3例行PET-CT,均考虑为恶性病变可能。实验室检查,血白细胞、血红蛋白、血小板、肌酐、C反应蛋白、乳酸脱氢酶、红细胞沉降率、IgM、IgG、IgA、CA242、CA125、CA199、癌胚抗原及尿蛋白均无明显异常。8例术前查24 h尿儿茶酚胺,除1例去甲肾上腺素升高(174.82 μg/24 h,正常值16.69~40.65 μg/24 h)外,其余7例均为正常水平,见表2。
1.2 方法
3例(例8、10、12)术前考虑为腹膜后Castleman病,给予激素治疗(口服泼尼松60~90 mg每日1次5、3、6天);3例术前诊断副神经节瘤,2例考虑为嗜铬细胞瘤,其余5例术前诊断腹膜后肿物,术前均未予特殊治疗。
12例行开腹或腹腔镜肿物切除术,1例行腋窝淋巴结活检术。
术后第1年每3个月复查一次,腹膜后彩超与腹盆腔CT交替进行。此后每6个月复查一次。2017年11月统一电话随访。
2 结果
1例(例12)因腹膜后及腋窝多发淋巴结肿大,且无单一肿物,故仅行腋窝淋巴结活检术,手术时间15 min,术后病理提示浆细胞型Castleman病。术后3天出院,口服沙利度胺100 mg每晚一次,规律随访,目前术后7个月,腹膜后淋巴结体积无明显改变。
其余12例行开腹(3例)或腹膜后腹腔镜(9例)肿物切除术,肿物均完整切除,手术时间45~210 min,平均144 min,出血量10~1500 ml,平均420 ml,术中见肿物与周围组织轻中度粘连,术后住院时间3~7 d,平均5.4 d,无明显并发症发生。12例大体病理(图2A)肿块边界清晰,包膜完整光滑,切面灰黄或灰粉,质地软~中,内部可见钙化或坏死,病理确诊为Castleman病,透明血管型10例(图2B),混合型1例,浆细胞型1例。12例术后均未行放化疗,随访1~58个月,平均25个月,1例失访(例2),1例(例6)术后半年发现原位复发,此后缓慢增大,目前术后27个月,肿瘤直径3 cm,无特殊不适,仍在密切随访,其余10例均恢复良好,无复发迹象。
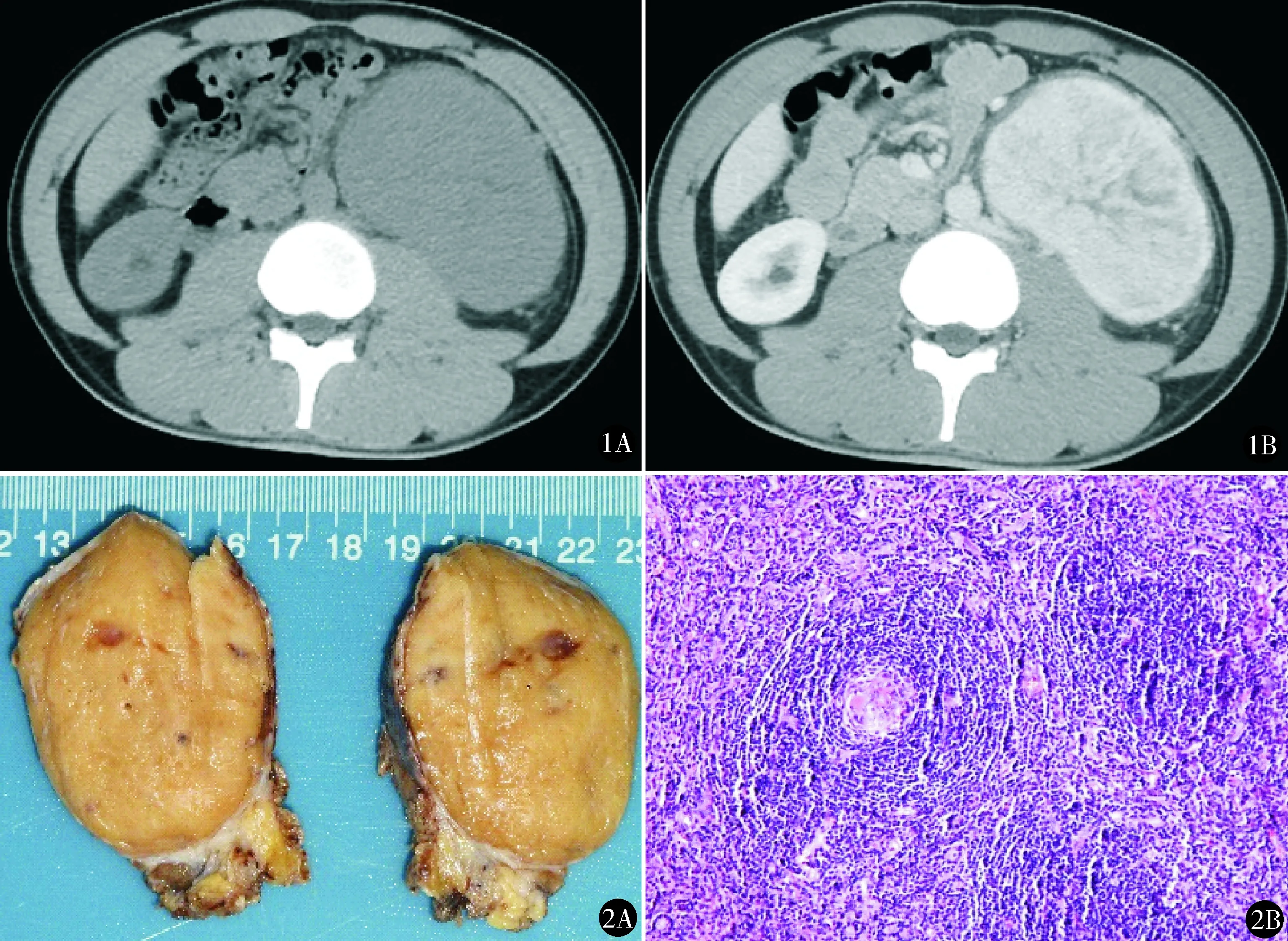
图1 例9,术前CT平扫(A)示左侧腹膜后软组织密度占位,11.8 cm×7.9 cm,动脉期(B)呈明显不均匀 图2 大体病理(A)切面成黄色,镜下(B)见滤泡树突状细胞按同心形排列呈“洋葱皮样”改变,诊断透明血管型(HE染色 ×200)
3 讨论
Castleman病又称血管滤泡性淋巴组织增生或巨大淋巴结病。是一种少见的原因未明的反应性淋巴结病。临床上可根据肿大淋巴结分布和器官受累的情况,分为单中心型(unicentric CD,UCD)和多中心型(multicentric CD,MCD)。单中心型发生率相对较高,美国每年新增Castleman病中,单中心型约占80%,常发生于20~30岁人群,累及单个淋巴结区域,症状较轻,预后较好[2];多中心型发生率相对较低,常发生于40~60岁人群,可累及多个淋巴结区域以及肝、肺、肾等重要脏器,多有全身症状,预后差[3]。腹膜后Castleman病少见。Talat等[4]包括404例的系统回顾表明,Castleman病最常累及的部位是胸(24%)、颈(20%)、腹(18%),腹膜后占位仅占14%。腹膜后Castleman病多为单中心型[5,6],虽然多中心型亦有累及腹膜后淋巴结可能,但极为少见。
Castleman病的发病机制尚不完全清楚,根据目前的研究结果[7],白细胞介素6(IL-6)异常表达和人疱疹病毒8(HHV-8)感染是较为公认的可能引起该病的原因。其中IL-6是与Castleman病发病关系最为密切的细胞因子之一。首先,早期研究显示Castleman病患者血清标本和淋巴结标本中IL-6表达上调[8];其次,单中心型患者通过手术切除病灶后,可观察到IL-6水平显著下降[9];第三,多中心型患者采用针对IL-6的靶向治疗后,发热、乏力等全身症状能够明显改善[4]。除此之外,推测系统性炎症性疾病、除IL-6外的其他细胞因子(包括白介素1、白介素5、白介素10、肿瘤坏死因子等)以及除HHV-8外的其他病毒(EB病毒等)感染可能与该病相关。
单中心型Castleman病通常无症状,往往是通过体格检查或影像学检查发现肿大淋巴结才引起关注。肿大的淋巴结中位直径约5.5 cm[4]。少见的情况下,病变的卡压和压迫效应(例如压迫邻近血管、气道)所导致的症状可能是患者就诊的原因[5]。与大部分单中心型Castleman病类似,本组12例单中心型腹膜后Castleman病也有7例是在影像学检查时意外发现病灶,少数患者受病变压迫产生腹部或腰背部疼痛的症状。多中心型Castleman病累及多个淋巴结区域(可包括腹膜后区域),随病变部位、病理类型不同,临床表现也复杂多变。本组1例多中心Castleman病患者因发热、四肢斑疹就诊。除此之外,多中心Castleman病还可以出现盗汗、体重减轻、虚弱或乏力等全身症状,甚至合并肺、肾等重要脏器受累。
腹膜后Castleman病可以合并皮疹、天疱疮等[6],临床上诊断为副肿瘤性天疱疮(paraneoplastic permphigus, PNP),十分罕见。PNP以黏膜损害,尤其是口腔黏膜广泛糜烂,皮肤多形性皮疹,且伴发肿瘤为特征。Kim等[10]报道,超过10%的PNP继发于Castleman病。PNP采用类固醇激素、免疫抑制剂等内科治疗无效,如不能及时诊断并及时将肿瘤切除,死亡率甚高。本组1例因术前口腔、外阴黏膜糜烂,周身红斑、水疱,合并腹膜后占位,诊断为副肿瘤性天疱疮,手术切除腹膜后病灶后病理证实Castleman病,目前随访21个月,局部无肿瘤复发,皮肤红斑的范围和黏膜糜烂的程度较术前缩小、好转。所以,对于疑似PNP者,应进一步行腹部及腹膜后B超、腹盆CT等检查,早期发现潜在的肿瘤。
腹膜后Castleman病大多数情况下常规实验室检查无明显异常,本组13例实验室检查均未见明显异常。但少数情况下,特别是多中心Castleman病可有贫血、血小板减少、红细胞沉降率增快、乳酸脱氢酶升高、低白蛋白血症、多克隆高丙种球蛋白血症、C反应蛋白和IL-6水平升高[4]。影像学检查方面,CT通常显示为边界清楚的软组织密度影,增强后可见均匀或非均匀强化;PET-CT可提示高摄取病灶。
需要强调的是,前述检查均仅有提示意义,诊断最终依赖病理。病理可分为透明血管型、浆细胞型和混合型三种类型:①透明血管型最多见,显微镜下可见异常的淋巴滤泡和萎缩或退化的生发中心,周围可见小淋巴细胞组成的宽阔覆盖区域。可见数根小血管穿入,血管内皮明显肿胀,管壁增厚,后期呈玻璃样改变。血管周围有数量不一的嗜酸性或透明状物质分布。还可见到2个或更多紧密相邻的萎缩生发中心被一个小淋巴细胞组成的覆盖区域包围。退化的生发中心通常呈透明样化,其内的淋巴细胞减少,主要由大量残余的滤泡树突状细胞组成。树突状细胞表达CD21、CD23、CD35和表皮生长因子受体。这些滤泡树突状细胞会产生按同心形排列呈“洋葱皮样”外观的典型形态学。②浆细胞型显微镜下可见增生性B细胞滤泡(生发中心),通常也有一些退化的滤泡。滤泡间区富含血供且可见成片的浆细胞。生发中心可见较为典型的反应性特征(核分裂象易见,包含细胞凋亡碎片的巨噬细胞等)。该型一般缺乏前述的“洋葱皮样”典型外观。③混合型兼具透明血管型和浆细胞型特征的混合组织学外观[11]。
由于腹膜后Castleman病行CT检查多表现为腹膜后局限性占位,动脉期强化较明显,甚至生长抑素受体显像亦为阳性表现,因此临床上主要需与嗜铬细胞瘤和副神经节瘤鉴别。但病史中,腹膜后Castleman病多无高血压发作,24 h尿儿茶酚胺基本为正常水平,MIBG显像均为阴性,且影像学检查常见肿物周围多发的肿大淋巴结影,此为Castleman病与嗜铬细胞瘤及副神经节瘤的鉴别要点。但临床中,静止性嗜铬细胞瘤及副神经节瘤也可有上述不典型表现。本组13例中,仅3例术前诊断Castleman病,5例考虑为嗜铬细胞瘤或副神经节瘤,可见二者的鉴别仍存在一定的难度,手术切除病灶后获得病理才是诊断的“金标准”。
单中心型Castleman病手术切除治疗效果良好。目前一般认为,对于包括腹膜后Castleman病在内的单中心型Castleman病,手术切除都是能够治愈疾病的“金标准”方案。而且,对于术前难以明确诊断的腹膜后Castleman病,手术不仅是治疗手段,也是最重要的确诊途径。具体手术方式可根据病灶的大小、范围、与周围组织关系等综合决定。对于部分不得不切除肾上腺者,术后可能需要激素替代治疗[5];对于手术难以完整切除病灶者,术后放疗可能有一定作用[12];对于手术困难病例,不必追求完全切除,部分切除亦有较好疗效(同时可帮助确诊)[13]。对于合并副肿瘤性天疱疮者,肿瘤切除后通常天疱疮会消退。多中心型Castleman病为多系统受累,故多不能受益于外科手术,激素治疗、化疗和放疗后有部分缓解。至今多中心型Castleman病尚缺乏“标准治疗”[14]。
由于病例极少,目前尚无专门针对腹膜后Castleman病预后的准确数据。目前最大规模的Castleman病回顾性研究[2]表明,大多数单中心型患者针对病灶进行完全切除后,可获得长期生存,5年生存率可高达97.1%。本组12例单中心Castleman病平均随访25个月,除1例失访,1例原位复发外,其余10例均恢复良好,且无复发迹象。尽管预后良好,仍然建议对此类患者进行规律随访,因为Castleman病可能导致淋巴瘤风险升高。Larroche等[15]报道6例Castleman病发生非霍奇金淋巴瘤。多中心型Castleman病由于多系统受累,治疗效果不满意,预后一般不良,5年死亡率高达35%[3],死亡原因主要包括疾病进展、感染、呼吸衰竭等。本组1例多中心Castleman病,目前随访7个月,恢复良好,腹膜后淋巴结体积无明显改变。
综上所述,腹膜后Castleman病多为单中心病变,多无明显临床症状。CT检查对诊断有一定帮助,但确诊仍靠病理。单中心Castleman病手术切除疗效好,术后可长期生存。多中心Castleman病更为罕见,临床表现多变,受累系统较多,治疗效果不佳。
